36 Cardiovascular Risk in Rheumatic Disease
For nearly half a century, excess rates of cardiovascular disease (CVD) have been reported among patients with inflammatory rheumatic diseases.1–5 More recently, the discovery of the inflammatory and immune mechanisms underlying atherosclerosis has spurred renewed interest in the association between CV risk and the rheumatic diseases. In this chapter we review the risks of cardiovascular comorbidity in the rheumatic diseases, focusing on rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). We also discuss the contribution of traditional and nontraditional cardiovascular risk factors to the observed excess CVD risk.
Cardiovascular Mortality
Rheumatoid Arthritis
The mortality of patients with established RA is known to be higher than that of the general population.6–10 Approximately 50% of all deaths in RA subjects are attributable to cardiovascular causes including ischemic heart disease (IHD) and stroke,11 and CVD appears to occur earlier in individuals with RA. The latter observation is consistent with the recent hypothesis of accelerated aging in RA in general.12 More than 50% of premature deaths in RA are due to CVD. A meta-analysis of 24 mortality studies in RA, published between 1970 and 2005, reported a weighted combined all-cause standardized mortality ratio (met-SMR) of 1.50 (95% confidence interval [CI],) with similar increases for IHD (met-SMR, 1.59; 95% CI, 1.46 to 1.73) and stroke (met-SMR, 1.52; 95% CI, 1.40 to 1.67); and for men (met-SMR, 1.45) and women (met-SMR, 1.58).13 Moreover, patients with RA frequently experience “silent” IHD and/or silent myocardial infarction (MI), showing no symptoms at all before a sudden cardiac death. Sudden cardiac deaths are almost twice as common in RA patients as in the general population (hazard ratio [HR], 1.99; 95% CI, 1.06 to 3.55).14
The excess CV mortality in RA may be confined to, or at least substantially higher in, subjects who are rheumatoid factor (RF) positive.3,15–18 The link may be even stronger with anticitrullinated protein antibody (ACPA) positivity.19 As might be expected, the relative risk of CV mortality is highest in younger age groups (those younger than 55 in whom controls have lower absolute risk and therefore in whom the distinction from those with RA is likely to be exacerbated) and in women, while the attributable risk is highest in the oldest age groups and in men.17,20,21
Controversy persists regarding how soon after symptom onset the excess CV mortality risk becomes apparent and/or whether there is a secular trend toward improving CV mortality in RA (as is seen in the general population). This may be partially explained by differences in the period of follow-up (i.e., follow-up starting from the time of symptom onset, from a physician’s diagnosis of RA, from the date of fulfillment of American College of Rheumatology (ACR) criteria, or other diagnostic criteria). The latter may not occur until some years after the first symptoms. In the Norfolk Arthritis Register (NOAR) the excess CV mortality is detectable beginning around 7 years after symptom onset.15 In a Dutch inception cohort of 1049 RA patients recruited between 1985 and 2007, excess mortality became apparent around 10 years after diagnosis (with all subjects having <1-year symptom duration).22 Unfortunately, there appears to be little evidence that CV mortality has improved significantly. A meta-analysis that included 17 studies (91,916 patients) reported CV mortality risk between 1976 and 2007, showing no trend toward improving CV SMR with time.23,24
Systemic Lupus Erythematosus
As first described by Urowitz and colleagues2 in 1976, mortality in SLE appears to follow a bimodal pattern, with an early peak in mortality (within 1 year of diagnosis) and a later peak occurring >5 years after diagnosis). Reported survival in the first 5 years of SLE has improved considerably from about 50% in the 1950s to more than 90% in the 1990s.25 However, there is some question as to whether this may be due, at least in part, to earlier diagnosis and improved ascertainment of mild cases. In the Toronto lupus cohort of 1241 patients recruited between 1970 and 2005, the SMR improved from 13.84 (range, 9.78 to 19.76) during 1970 to 1978 for those who entered the cohort in that decade to 3.81 (range, 1.98 to 7.32) during 1997 to 2005 in those who entered the cohort in that time period.26 The SMR during 1997 to 2005 was similar for patients regardless of their disease duration, ranging from 3.23 for those who had entered the cohort in 1970 to 1978 to 3.93 for those who had entered the cohort in 1988 to 1996. Similarly, evidence from Olmsted County, Minnesota showed an SMR of 2.70 with significant improvement in survival in recent decades.27 In a study of 434 female lupus patients from Seoul, Korea followed from 1992 to 2002, the SMR was 3.02 (95% CI, 1.45 to 5.55).28 No study has been large enough to permit study of cause-specific SMR.
Cardiovascular Comorbidity
Rheumatoid Arthritis
Ischemic Heart Disease
Patients with RA are at increased risk of IHD.5,14,20,24,29–32 Data from the Rochester Epidemiology Project have shown that, in the 2-year period immediately preceding the fulfillment of the ACR 1987 criteria, RA patients were more likely to experience hospitalization for MI (odds ratio [OR], 3.17; 95% CI, 1.16 to 8.68) and unrecognized (“silent”) MI (OR, 5.86; 95% CI, 1.29 to 26.64) than age- and sex-matched controls. The increased risk of unrecognized MI persisted after the diagnosis of RA (HR, 2.13; 95% CI, 1.13 to 4.03). Holmqvist and colleagues failed to demonstrate a statistically significant elevated increase in MI, angina, or heart failure before the onset of symptoms in two large Swedish cohorts,33 although trends toward such elevation were reported. As in studies of mortality, these results suggest that accelerated atherosclerosis begins at the onset of RA symptoms, or even earlier, and not at the time of diagnosis or later in the disease course.
Patterns of clinical care and outcome after MI may vary in persons with RA when compared with the general population. Some evidence suggests that although RA patients receive similar MI care to non-RA patients, they experience higher rates of heart failure and death after MI.32,34,35 However, others have recently reported that RA patients who experience acute MI receive acute reperfusion and secondary prevention medications (including β-blockers and lipid-lowering agents) less frequently than controls.36 Moreover, another group recently reported that among patients with MI, those with RA were more likely to undergo thrombolysis and percutaneous coronary intervention (PCI) but less likely to receive medical therapy or coronary artery bypass grafting, or both.37 That study also suggested that patients with RA may have an in-hospital survival advantage, particularly those undergoing medical therapy and PCI, though potential confounding could not be ruled out.
Heart Failure
Patients with RA are also at increased risk of developing heart failure compared with the general population.38,39 In the Rochester RA cohort, the cumulative incidence of congestive heart failure (CHF; defined according to the Framingham criteria) at 30-year follow-up was 34% compared with 25% in the non-RA cohort (Figure 36-1; P < 0.001). Even after adjustment for demographics, CV risk factors, and IHD, patients with RA had almost twice the risk of developing CHF as non-RA subjects (HR, 1.87; 95% CI, 1.47 to 2.39). This increased risk of CHF appeared to be predominantly in the subgroup of RA patients who were RF positive (HR in RF-positive patients, 2.59; 95% CI, 1.95 to 3.43 vs. 1.28 in RF-negative patients; 95% CI, 0.93 to 1.78).
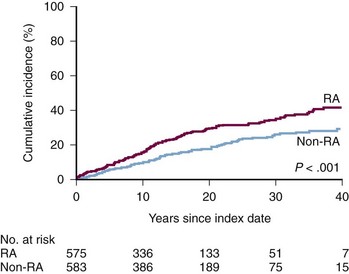
The clinical presentation of CHF in patients with RA differs from CHF in non-RA patients.40 RA patients with CHF are less likely to be obese, be hypertensive, or have clinical IHD. Moreover, RA patients with CHF are less likely to have typical signs and symptoms. Importantly, the proportion of CHF patients with preserved ejection fraction (>50%) is significantly higher among RA compared with non-RA patients.40 It has also been shown that RA patients with CHF tend to be investigated and managed less aggressively.40 Finally, RA patients with CHF also appeared to have poorer outcomes, experiencing approximately twice the risk for death in the period immediately after detection of heart failure compared with non-RA patients.41
Systemic Lupus Erythematosus
Ischemic Heart Disease
Accelerated atherosclerosis is an established complication of SLE.30,42–49 The prevalence of atherosclerotic vascular events varies from 1.8% in early disease to more than 27% later in the course of SLE.47,50–52 The majority of studies reported increased risk of MI ranging from 2- to greater than 10-fold in various SLE patient groups compared with the general population.30,45,47,53 This increased relative risk of MI is particularly apparent in younger SLE patients. The most striking example comes from the University of Pittsburgh lupus cohort, where women with SLE aged 35 to 44 were more than 50 times as likely to have an MI compared with women without SLE in the Framingham Offspring study (relative risk [RR], 52.43; 95% CI, 21.6 to 98.5).47 Furthermore, the majority (67%) of women with SLE were younger than 55 years of age at the time of their first cardiac event. In addition, a greater than twofold increased risk of hospitalizations for MI has been reported in young women with SLE between 18 and 44 years of age compared with those without SLE (OR, 2.27; 95%CI, 1.08 to 3.46).49
A recent study of patients undergoing coronary revascularization procedures found no significant differences in the mean percent of coronary stenosis and total occlusion in SLE versus non-SLE subjects.54 Except for the increased likelihood of lesions confined to the left anterior descending artery in SLE versus non-SLE subjects (42.3% vs. 19.3%; P = 0.003), the pattern of coronary involvement including artery dominance and prevalence of multivessel disease appeared similar in SLE versus non-SLE subjects. However, the study reported significantly worse cardiovascular outcomes at 1 year following PCI in SLE versus non-SLE subjects including higher risk of MI (15.6% vs. 4.8%; P = 0.01), and repeat PCI (31.3% vs. 11.8%; P = 0.009) in SLE, even after adjustment for important co-variates.54,55 Given increased vulnerability of atherosclerotic plaque in SLE, which is associated with the risk of occlusive events irrespective of size of the plaque, these findings suggest an increased risk of unfavorable cardiovascular events in SLE patients versus non-SLE subjects with a seemingly similar pattern of coronary involvement.56
In a large population-based study of patients hospitalized in California with acute MI from 1996 to 2000, in-hospital mortality and length of stay were essentially similar in patients with SLE compared with those who did not have SLE adjusting for age, race, ethnicity, type of medical insurance, and Charlson Index. In contrast, data from the 1993 to 2002 U.S. Nationwide Inpatient Sample showed significantly increased rates of in-hospital mortality (RR, 1.46; 95% CI, 1.31 to 1.61) and prolonged hospitalization (RR, 1.68; 95% CI, 1.43 to 2.04) for acute MI in SLE compared with controls, adjusting for age, sex, race/ethnicity, income, and CHF.55 Some differences in methodology (i.e., smaller sample size and older age of SLE patients and control subjects in the earlier study and shorter observation period in the later study) may at least in part explain these contrasting results. Considering the presence of myocardial involvement, chronic systemic inflammation, vasculitis, and hyperviscosity syndrome in SLE, worse outcomes following acute coronary events and associated interventions can reasonably be expected.57 However, this requires further study.
Heart Failure
The risk of CHF and related hospitalization in SLE appears to be substantially increased.49,55,58 In particular, young women with SLE between 18 and 44 years of age have a greater than 2.5-fold increased risk of hospitalization for CHF versus those who did not have SLE, even after adjustment for age, race, insurance status, hospital characteristics, and the presence of hypertension, diabetes mellitus, and chronic renal failure.49 Consequently, CHF accounted for a substantially higher percent of hospitalizations in women with SLE versus those without SLE within this age group (1.32% vs. 0.35%, respectively; P < 0.0001). The nature of CHF in SLE is likely multifactorial, only partly attributable to atherosclerosis.52,59,60 The presentation of CHF in SLE may vary from severe overt CHF to insidious myocardial involvement.59–63 Finally, mortality in SLE patients with CHF is significantly higher than in those without CHF (17.9% vs. 5.8%; P < 0.001) and approximates 3.5-fold as compared with the general population.55,59
Traditional Risk Factors for Cardiovascular Disease
Traditional Cardiovascular Risk Factors
Smoking in Rheumatoid Arthritis
Smoking is a known risk factor for the development of RA, in particular RF and ACPA-positive RA. Thus as expected, there is a higher prevalence of current smokers and ex-smokers among subjects with RA than among the general population. In a meta-analysis of four case-control studies (1415 RA patients) of traditional CVD risk factors in RA, the prevalence of smoking was found to be significantly higher than in controls (OR, 1.56; 95% CI, 1.35 to 1.80).64 Smokers with RA also appear to have a worse prognosis in terms of RF titers, disability, radiographic damage, and treatment response.65 There is a known interaction among smoking, human leukocyte antigen (HLA) DR1 shared epitope (SE) alleles, and the production of ACPA66 and among smoking, ACPA, and the SE in premature CVD mortality in RA.19
Smoking in Systemic Lupus Erythematosus
In a case-control study involving 250 women with SLE, smoking was no more common than in the general population (RR, 0.86; 95% CI, 0.59 to 1.24).67 Nevertheless, smoking was a risk factor for vascular events in the cohort overall and in the inception subcohort.51
Hypertension in Rheumatoid Arthritis
Hypertension is common in patients with RA, but it remains unclear whether it is more common than in the general population. A recent meta-analysis of seven case-control studies (1053 RA patients) found the prevalence of hypertension to be the same in RA patients as in controls (OR, 1.09; 95% CI, 0.91 to 1.31).64 There is, however, some evidence for underdiagnosis and undertreatment of hypertension in RA patients.68 Multiple other factors may influence blood pressure control in persons with RA including physical inactivity, obesity, specific genetic polymorphisms, and several antirheumatic medications including nonsteroidal anti-inflammatory drugs (NSAIDs), corticosteroids, leflunomide, and cyclosporine.
Hypertension in Systemic Lupus Erythematosus
Hypertension is substantially more common in women with lupus than in the general population. A case-cohort control study found the relative risk of hypertension in women to be 2.59 (95% CI, 1.79 to 3.75).67 Hypertension has been noted to be a predictor of mortality and vascular events in lupus in a number of studies.50,69 Hypertension was found to be a predictor of vascular events in the total Toronto cohort of 1067 patients but not in the 561 patients in the inception cohort.51
Dyslipidemia in Rheumatoid Arthritis
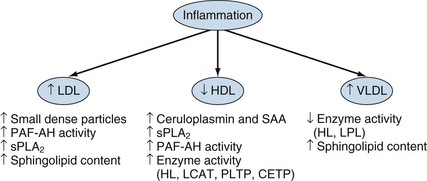
Although findings have been somewhat inconsistent, hyperlipidemia appears to have a paradoxical relationship with CV risk in persons with RA,70–74 namely decreased lipid levels associated with increased CV risk. Serum levels of total cholesterol and low-density lipoprotein (LDL) cholesterol decline precipitously during the 3- to 5-year period before RA incidence,75 and lower total and LDL cholesterol levels have been shown to be associated with higher CV risk.76 Suppression of total and LDL cholesterol levels during acute or chronic high-grade inflammation is well described, as is a proportionately greater suppression of high-density lipoprotein (HDL) cholesterol, resulting in a disadvantageous atherogenic index (total-to-HDL cholesterol ratio).77 This may explain the fact that hyperlipidemia (high total or LDL cholesterol) appears to be less common in RA compared with non-RA subjects.70,78,79 Dyslipidemia (alterations of individual lipid components and their ratios as defined by specific criteria) may affect up to half of all RA patients in hospital care.80 A recent meta-analysis showed that RA is associated with an abnormal lipid pattern, principally low levels of HDL cholesterol.81 Lipid alterations appear to predate the diagnosis of RA. Serum levels of total cholesterol and LDL cholesterol decline precipitously during the 3- to 5-year period before RA diagnosis.75,82 In vitro animal model and human in vivo studies in subjects without RA clearly demonstrate that the interplay between inflammation and lipid components is far more complex than simple alterations of their serum levels83 (Figure 36-2). For example, acute phase proteins such as serum amyloid A and phospholipase A2 can alter HDL composition and function, whereas inflammation may have profound effects on enzymes fundamental to the metabolism of HDL (e.g., hepatic lipase) or indeed the enzymatic content of HDL itself (e.g., reduced paraoxonase); this may increase susceptibility to oxidation and convert HDL to a more pro-oxidant, pro-atherogenic complex. Such inflammation-induced alterations of structure and function are not confined to HDL but also involve triglycerides and LDL; they require further study, specifically in RA and in the context of disease control through nonbiologic and biologic disease-modifying antirheumatic drugs (DMARDs).84 To date, several studies suggest antirheumatic therapy mediates effects on lipid levels including glucocorticoids, hydroxychloroquine, gold, cyclosporine, and the biologics anti–tumor necrosis factor (TNF), rituximab, and tocilizumab: these are generally short-term studies of small numbers of patients addressing predominantly serum levels rather than other modifications or mechanisms.83,85 Multiple other factors are involved in lipid regulation and function including physical activity, adiposity, diet, alcohol intake, and smoking. However, their effects have not been assessed in any detail in persons with RA. Similarly, the importance of genetic regulation of lipid metabolism, particularly in the context of gene-environment interactions, has not been addressed in the RA population. This may be particularly important because lipid alterations appear to predate the diagnosis of RA.
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


