96 Calcium Crystal Disease
Calcium Pyrophosphate Dihydrate and Basic Calcium Phosphate

Acr and EULAR Criteria for Disease
 Supplemental images available on the Expert Consult Premium Edition website.
Supplemental images available on the Expert Consult Premium Edition website.
Table 96-1 features proposed diagnostic criteria for calcium pyrophosphate dihydrate (CPPD) deposition disease, adapted by the author from the proposed European League Against Rheumatism (EULAR) criteria1 and the original criteria proposed in the past by Daniel J McCarty and colleagues. The diagnosis is based on detection of CPPD crystals by one or more methods. These include not only standard clinically applied radiography but also high-resolution ultrasound to detect hyaline articular cartilage or fibrocartilage calcifications characteristic of CPPD crystal deposition (termed chondrocalcinosis). However, identification of CPPD crystals via compensated polarized light microscopic analysis of synovial fluid in the absence of joint infection or other cause of arthritis is the gold standard for diagnosis of CPPD crystal deposition disease, particularly for acute CPPD crystal-associated arthritis (termed pseudogout). Detection of typical CPPD crystals in tissue sections also can be accomplished whether specimens are fixed in formaldehyde or ethanol, unlike the case for monosodium urate crystals (which dissolve in formaldehyde). On occasion, specialized crystal analytic approaches including x-ray energy spectroscopy and powder diffraction analysis or atomic force microscopy may be helpful in establishing or confirming CPPD crystal deposition. In assessing the form of deposited crystals in calcifications, determination of the calcium/phosphate ratios and the spacing of x-ray powder diffraction lines provide the most specific information.
Table 96-1 Proposed Diagnostic Criteria for Calcium Pyrophosphate Dihydrate (CPPD) Crystal Deposition Disease
Modified from McCarty DJ: Crystals and arthritis, Dis Month 6:255, 1994.
Epidemiology
Key Points
Chondrocalcinosis including asymptomatic disease increases progressively in prevalence with aging.
In the past, studies of the prevalence of both CPPD crystal deposition disease2–5 and various forms of articular BCP crystal deposition disease have been predominantly based on characteristic plain radiographic features of the disease in limited numbers of joints. This is an incompletely sensitive and specific approach.2 Other studies have been based on results of synovial fluid analyses, but definitive studies based on pathologic findings on examination of articular cartilages, or imaging approaches superior to plain radiography, have not been done. As such, the true prevalence of both CPPD crystal deposition disease and pathologic articular BCP crystal deposition is not known.
It is clear that chondrocalcinosis including asymptomatic disease increases progressively in prevalence with aging.2–5 Idiopathic/sporadic chondrocalcinosis is rare before age 55, particularly in the absence of a history of joint trauma or knee meniscectomy. Studies of prevalence based on radiographs have estimated higher prevalence of chondrocalcinosis when the hands, wrists, pelvis, and knees have been surveyed. Indeed, most elderly patients with chondrocalcinosis of the knee also have detectable chondrocalcinosis in other joints. Knee meniscal fibrocartilage calcification alone has been detected in 16% of women aged 80 to 89 and in 30% of women older than 89,6 figures comparable with those obtained in other studies.7 In a radiographic survey study of hands, wrists, pelvis, and knees of patients admitted to a geriatrics ward, there was a 44% prevalence of chondrocalcinosis in patients older than 84, a 36% prevalence in the 75- to 84-year-olds, and a prevalence of 15% in 65- to 74-year-olds8; studies of U.K. and Italian community cohorts have been limited to analyses of fewer regions (the knee, or knee and pelvis, respectively) and have yielded lower numbers for prevalence.2–5
In a large U.K. community study the age-, sex-, and knee pain–adjusted prevalence of knee chondrocalcinosis was 4.5% for those older than age 40.3 In some studies, women have appeared somewhat more commonly affected by CPPD crystal deposition disease than men,1 but in the recent U.K. study, there was no sex predisposition, although strong association between osteoarthritis (OA) and chondrocalcinosis was confirmed.3 This appeared to be linked more to the presence of osteophytes rather than joint space narrowing with OA. Interestingly, an association between chondrocalcinosis and diuretic use was uncovered, proposed to be due to the capacity of diuretics to induce hypomagnesemia.3
CPPD crystal deposition disease is not uniform in epidemiology between populations. In a random sample of Beijing residents older than 60 years of age, radiographic chondrocalcinosis was compared with whites in the American Framingham OA Study.9 Chinese had a much lower prevalence of knee chondrocalcinosis, and wrist chondrocalcinosis was particularly rare in Chinese elderly.9 These findings were unexpected because there is an excess of knee OA in Beijing, and chondrocalcinosis and OA are quite commonly associated in the knee joint.
Genetics
The vast majority of CPPD crystal deposition disease is idiopathic/sporadic, but early-onset (defined as onset before age 55) familial disease also occurs.10 Two major chromosomal linkages, 8q and 5p, have been identified in studies of familial CPPD deposition disease. Linkage with chromosome 8q of both early-onset OA and chondrocalcinosis was given the designation CCAL1, but chromosome 5p-linked chondrocalcinosis (CCAL2) is broadly distributed and has been studied in greater detail than 8q chondrocalcinosis.10–12 Linkage of familial CPPD crystal deposition disease to the gene ANKH on chromosome 5p (which encodes a transmembrane protein with inorganic pyrophosphate [PPi] transport and other apparent functions discussed later) has been established in these studies.10–12 A search for ANKH mutation in 95 subjects with sporadic chondrocalcinosis uncovered a unique mutation (ΔE590) in one subject.11 Homozygosity for a single nucleotide substitution (−4 G to A) in the ANKH 5′-untranslated region that promotes increased ANKH messenger RNA expression was present in approximately 4% of British subjects previously thought to have idiopathic/sporadic chondrocalcinosis of aging.13
Familial chondrocalcinosis is heterogeneous, and, as one example, prominent CPPD and hydroxyapatite (HA) crystal deposits and cartilage and periarticular calcifications in association with OA were described in a kindred not yet linked to a specific chromosomal locus.14 A syndrome of spondyloepiphyseal dysplasia tarda, brachydactyly, precocious OA, and intra-articular calcifications with CPPD and/or HA crystals, as well as periarticular calcifications, was linked to mutation of the procollagen type II gene in indigenous natives of the Chiloe Island region of Chile.15 This population has a high prevalence of familial CPPD deposition disease. Families affected with diffuse idiopathic skeletal hyperostosis (DISH) and/or chondrocalcinosis have been identified in the Azores Islands, possibly reflecting an unidentified, shared pathogenic mechanism.16
Pathogenesis
Inflammatory conditions also may result from deposition of HA, as well as the closely related BCP crystals, octacalcium phosphate (OCP) (Ca8H2[PO4]6·5H2O; calcium-to-phosphate ratio, 1.33) and tricalcium phosphate or “whitlockite” (Ca3[PO4]2; calcium-to-phosphate ratio, 1.5) in periarticular structures such as the rotator cuff (calcific tendinitis) and subacromial bursa of the shoulder (see Chapter 46). CPPD and BCP crystal deposition, reviewed here, are by far the most prevalent arthropathies associated with calcium-containing crystals. Articular calcium oxalate crystal deposition is less common.
Articular cartilage, unlike growth plate cartilage, is specialized to avoid the process of matrix calcification. However, the matrix of articular hyaline cartilage, like that of fibrocartilaginous menisci, lends itself well to pathologic calcification,17 particularly in association with certain changes in extracellular matrix composition and hydration in aging and OA.18 Joint cartilage calcification reflects complex interplay between organic and inorganic biochemistry, ion transport, aging, genetics, inflammation, oxidative stress, and dysregulated chondrocyte growth factor responsiveness and differentiation. Pathologic cartilage calcification can reflect deficiencies of certain physiologic calcification inhibitors or upregulation of mediators that actively drive certain patterns of tissue injury culminating in calcification within degenerating cartilage.19,20
Alteration of the concentrations of calcium, inorganic phosphate (Pi,), PPi, and the solubility products of these ions are clearly at work in promoting CPPD and BCP crystal formation.19 The levels of ambient magnesium and the composition of the chondrocyte extracellular matrix influence the dynamics of CPPD crystal formation and help to determine whether predominantly monoclinic or triclinic CPPD crystals are predominantly formed.21,22 Significantly, monoclinic CPPD crystals are more inflammatory than triclinic CPPD crystals.23 Matrix effects for CPPD and BCP crystals, studied in experimental gel systems, include promotion of CPPD formation by adenosine triphosphate (ATP) and corticosteroids in conjunction with matrix type I collagen and osteopontin, whereas type II collagen and intact proteoglycans appear to suppress ATP-driven CPPD crystal formation in vitro.21,22 Experimental systems to analyze CPPD and BCP crystal deposition have commonly employed isolated matrix vesicle cell fragments from chondrocytes that are enriched in promineralizing constituents and provide a nidus for intiation of calcification, specifically with BCP crystals.21 Matrix vesicles are important in cartilage growth plate calcification, but it is not yet clear whether CPPD and BCP crystal formation in articular cartilages is driven more by matrix vesicle–mediated effects or nucleation of crystals in association with changes in extracellular matrix constituents, or both pathways. However, CPPD crystals are too large (micron size) to form within matrix vesicles. It should be noted that loci of pericellular concentration of PPi may be necessary to drive CPPD crystal formation at low micromolar PPi concentrations developing in cartilages with chondrocalcinosis.
Besides physical effects of calcium, Pi, and PPi on crystal nucleation and propagation, these same solutes exert a variety of mineralization-regulating effects on gene expression, differentiation, and viability in chondrocytes, mediated partly by calcium-sensing receptors and sodium-dependent inorganic phosphate co-transport in chondrocytes.24–27 Noxious effects of excess PPi on chondrocytes including induction of matrix metalloproteinase-13 (MMP-13) expression28 and promotion of apoptosis29 support the clinical terminology pyrophosphate arthropathy to describe chronic cartilage degenerative manifestations of CPPD crystal deposition disease.
Dysregulated Inorganic Pyrophosphate Metabolism in Pathologic Articular Cartilage Calcification
PPi is a potent inhibitor of the nucleation and propagation of BCP crystals.19 Concordantly, maintenance of physiologic extracellular PPi levels by chondrocytes and certain other cells serves to suppress calcification with HA, as illustrated in mouse models of deficient PPi generation and transport,25 and a variant of human infantile arterial calcification associated with periarticular calcification.30 The relatively unique capacity of chondrocytes to produce copious amounts of extracellular PPi is double-edged (Figure 96-1), as supersaturation of cartilage extracellular matrix with PPi is a major factor in promoting CPPD crystal deposition.19,31,32 Furthermore, excess PPi generation can promote BCP crystal deposition by providing a source for increased extracellular Pi generation via PPi hydrolysis by the ecto-enzyme tissue-nonspecific alkaline phosphatase (TNAP)19,25 (see Figure 96-1). Depending on cartilage ATP and PPi concentrations, as well as the level of activity of Pi-generating ATPases and TNAP, CPPD and HA crystal formation may be jointly promoted in cartilage, an event that commonly occurs clinically in OA.
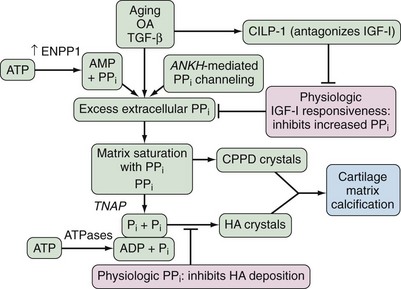
Role of ENPP1 and ANKH in Inorganic Pyrophosphate Metabolism in Chondrocalcinosis
Sporadic aging-associated CPPD crystal deposition disease is consistently linked with excess chondrocyte PPi-generating nucleotide pyrophosphatase phosphodiesterase (NPP) activity and augmented PPi generation by chondrocytes.19,31,32 In this context, the NPP family isoenzymes ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) (formerly known as NPP1 and plasma cell membrane glycoprotein-1 [PC-1]) and ENPP3 (formerly known as B10) actively generate PPi by hydrolysis of nucleoside triphosphates including ATP.19,31,32 ENPP1 plays a central role in sustaining and augmenting extracellular PPi in chondrocytes and certain other cells (see Figure 96-1). A substantial portion of ATP used by chondrocytes to generate extracellular PPi is provided by the mitochondria.19
ENPP1 is one of a family of enzymes that share NPP catalytic activity and modular type II transmembrane ecto-enzyme structures.19,33 ENPP1 plays by far the greatest role in augmenting extracellular PPi in chondrocytes.31,32 Significantly, marked and total ENPP1 deficiency states in vivo and in vitro are associated with up to 50% less plasma and extracellular PPi.24,30 In contrast, in idiopathic chondrocalcinosis, cartilage NPP activity and PPi levels may average approximately double those of normal subjects.34
Increased ENPP1 expression is associated with both calcification and apoptosis in degenerative human cartilages.31 In more advanced osteoarthritis, decreased cartilage ENPP1 has been described and promotes BCP crystal deposition.34a Direct upregulation of ENPP1 in chondrocytic cells stimulates calcification, as well as apoptosis.35 These effects are not shared by ENPP3, which likely has other intracellular “housekeeping” functions in chondrocytes.31 ENPP2, which is also expressed in normal cartilages, functions more actively in physiology as a lysophospholipase D, and ENPP2 only modestly stimulates chondrocytes to calcify in vitro.31
ANKH encodes a multiple-pass transmembrane protein that functions in PPi channeling36–39 (Figure 96-2) and possibly ATP release40 and regulation of Pi metabolism and uptake by the type III sodium-dependent Pi co-transporter Pit-1.41 ANKH promotes bidirectional movement of PPi at the plasma membrane in vitro,38 but the gradient for ANKH-stimulated PPi movement in chondrocytes (which generate abundant PPi both by their robust ENPP1 expression and intense matrix biosynthetic activity) is from the intracellular to the extracellular space.19 Indeed, ANKH transport of PPi generated intracellularly by ENPP128 may be the primary means to regulate extracellular PPi levels.19 Modeling of the PPi channeling function of ANKH has proposed 10 or 12 membrane-spanning domains in ANKH with an alternating inside/out orientation and with a central channel to accommodate the passage of PPi36,38 (see Figure 96-2).
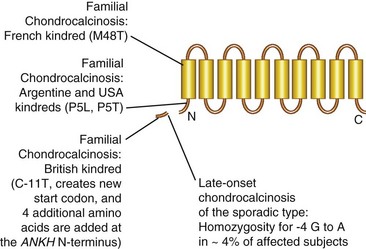
ANKH is clearly implicated in the pathogenesis of familial and idiopathic/sporadic chondrocalcinosis,10,13 and increased ANKH expression in cartilage is a factor in secondary chondrocalcinosis in OA28 (see Figure 96-1). In this context, expression of wild-type ANKH is highly regulated and ANKH is increased in OA and chondrocalcinotic cartilages.28 Interestingly, hypoxia, via effects of the transcription factor hypoxia inducible factor-1α, suppresses ANKH expression.42 It is conceivable that increased permeability to oxygen of fibrillated and fissured cartilage in OA favors increased ANKH expression. ANKH, in conjunction with signaling via extracellular Pi likely derived from PPi, promotes chondrocyte maturation to the procalcifying hypertrophic differentiation state.43 Figure 96-1 presents a model in which secondary alterations in chondrocyte expression of both wild-type ANKH and ENPP1 drive PPi supersaturation in cartilage in idiopathic/sporadic and OA-associated CPPD crystal deposition disease.
Mutations at different locations in ANKH can affect function and the skeleton in a manner including autosomal dominant chondrocalcinosis10,37–39 and certain other phenotypes. These include murine progressive ankylosis in the ank/ank mouse and human craniometaphyseal dysplasia associated with apparent decrease of the capacity to transport PPi within bone and effects on bone resorption and remodeling, putatively mediated in part by direct and indirect effects on osteoclasts.36,39,44 In a consanguineous family with mental retardation, deafness, and ankylosis, with painful small joint soft tissue calcifications, progressive spondyloarthropathy, osteopenia, and mild hypophosphatemia, the homozygous ANK missense mutation L244S was detected in all patients.45 The mutant ANK protein was expressed and localized to the plasma membrane, but fibrosis and mineralization of articular soft tissues developed in homozygotes, with heterozygous carriers of the L244S mutation showing mild osteoarthritis without metabolic alterations.45
Clinical heterogeneity even for chondrocalcinosis associated with ANKH mutations10 suggests differing functional effects of ANKH mediated by specific regions of the molecule. All the N-terminally clustered ANKH mutations identified to cause familial chondrocalcinosis (see Figure 96-2) appear to increase PPi transport.38 However, some ANKH mutations have distinct effects on chondrocyte differentiation.13 The M48T ANKH mutant in the French kindred appears functionally unique by association with increased intracellular PPi46 and also interrupts the interaction of ANKH with the sodium/phosphate co-transporter Pit-1.47 This may be functionally significant because elevated Pi increases both ANKH and Pit-1 expression and because ANKH and Pit-1 co-localize in chondrocytes.47 In addition are the Pi effects on chondrocyte differentiation discussed later. In 5p familial chondrocalcinosis, subtle gain of function of intrinsic ANKH PPi channeling activity may lead to chronic, low-grade chondrocyte “PPi leakiness,” thereby causing matrix supersaturation with PPi, CPPD crystal deposition, and cartilage degeneration.10,37,48 An alternative mechanism of disrupting PPi metabolism may be promoted by the ANKH mutation ΔE590 linked with a case of sporadic chondrocalcinosis11 because ANKHΔE590 appears to indirectly suppress PPi catabolism by association with impairing TNAP expression.49
Effects of Imbalance of Chondrocyte Growth Factor Responses on Inorganic Pyrophosphate Metabolism in Chondrocalcinosis
The chondrocyte growth factor transforming growth factor (TGF)-β stimulates ATP release by chondrocytes,40 as well as ENPP1 expression and ENPP1 subcellular movement to the plasma membrane, which drive elevation of extracellular PPi.32,50 Interleukin (IL)-1β suppresses both ENPP1 expression and extracellular PPi in chondrocytes and blocks the effects of TGF-β on PPI.32,50 The capacity of TGF-β to raise chondrocyte PPi rises with aging, as does TGF-β–stimulated NPP activity,51 whereas growth-promoting effects of TGF-β decrease with aging in articular chondrocytes.52 The anabolic chondrocyte growth factor insulin-like growth factor-I (IGF-I) normally suppresses extracellular PPi (as well as ATP release)40 in chondrocytes53 (see Figure 96-1). Moreover, chondrocyte IGF-I resistance is characteristic of OA and aging cartilages54 (see Figure 96-1). IGF-I induces expression of cartilage intermediate layer protein (CILP) (see Figure 96-1), a secreted cartilage matrix molecule. CILP’s expression rises in aging and OA and is most abundant in the middle zone of articular cartilage where CPPD crystal deposition is most prevalent. Significantly, the CILP-1, but not the CILP-2 isoform, promotes increased extracellular PPi in chondrocytes indirectly by antagonizing IGF-I at the receptor level.54
CPPD Deposition Disease Secondary to Primary Metabolic Disorders: Relationship to Inorganic Pyrophosphate Metabolism and Chondrocyte Differentiation
Hypophosphatasia, hypomagnesemic conditions (including the Gitelman’s variant of Bartter’s syndrome), hemochromatosis, and hyperparathyroidism are the best-characterized primary metabolic disorders linked to secondary CPPD crystal deposition disease.55 Increased joint fluid PPi levels in each of these conditions suggests at least one common thread in the pathogenesis of chondrocalcinosis via cartilage PPi excess.56 Magnesium is a cofactor for pyrophosphatase activity, and iron excess can suppress pyrophosphatase activity. Hypercalcemia may promote CPPD crystal deposition in hyperparathyroidism (and in familial hypocalciuric hypercalcemia)57 by effects beyond cartilage matrix supersaturation with ionized calcium such as calcium function as a cofactor in ENPP1 catalytic activity, as well as chondrocyte-activating effects mediated by the calcium-sensing receptor.27 In addition, normal articular chondrocytes express parathyroid hormone/parathyroid-hormone-related protein (PTH/PTHrP) receptors, and functional responses of chondrocytes to PTH can promote proliferation, altered matrix synthesis, and mineralization.58,59
Hypophosphatasia is due to deficient activity of TNAP, consequently with effects including limitation of hydrolysis PPi to generate Pi.25 TNAP is a major physiologic antagonist of ENPP1-mediated elevation of extracellular PPi.25 Conversely, physiologic ENPP1-induced PPi generation antagonizes the essential promineralizing effects of TNAP mediated by Pi generation,25 and cartilage PPi excess presumably drives chondrocalcinosis in hypophosphatasia. Enpp1 knockout mice and mice homozygous for the ENPP1 truncation mutant ttw demonstrate marked articular cartilage calcification with HA and OA, as well as ankylosing spinal ligament hyperostosis and synovial joint ossific fusion; extracellular PPi levels and mineralization disturbances in soft tissues (but not long bones) of Enpp1 knockout and TNAP-deficient mice are mutually corrected by cross-breeding.25
Inflammation, Hypertrophic Chondrocyte Differentiation, and Transglutaminase 2 in Joint Cartilage Calcification
Regulated changes in chondrocyte differentiation and viability appear to be a mechanistically unified process that promotes joint cartilage HA and CPPD crystal deposition, as well as OA.60 Such changes include development of foci of chondrocyte maturation to hypertrophy,60 with the presence of hypertrophy, as seen in histopathology of the knee cartilage in Figure 96-3, and apoptosis of chondrocytes typically found adjacent to cartilage calcifications.61,62 Articular chondrocyte hypertrophy60 is associated with heightened PPi generation; increased production of calcifying, membrane-limited cell fragments known as matrix vesicles63; and certain other calcification-promoting changes in differentiation including alteration of extracellular matrix composition, such that osteopontin (which promotes CPPD crystal formation) is increased and normal collagen subtype composition in the matrix is lost.64 Altered TGF-β signal transduction in aging and OA may be involved in promoting chondrocyte hypertrophy.65
Pi taken up by Pit-1 sodium-dependent co-transport and calcium sensing can modulate chondrocyte hypertrophic differentiation and apoptosis, as well as PPi-modulating responses to TGF-β.19,31,66–69 Local upregulation of PTHrP expression also may be one of the shared features driving sequential chondrocyte proliferation and altered differentiation in growth plate chondrocytes and articular chondrocytes.27 Chondrocyte apoptosis also promotes calcification partly through the calcifying potential of apoptotic bodies functioning as “inside-out” matrix vesicles on release from dying chondrocytes.70–72 Mitochondrial dysfunction, a central factor in tissue aging and an apparent mediator of OA progression in aging,73,74 also can stimulate cartilage matrix degeneration and calcification. Mitochondria are remarkably specialized to regulate calcification, and apoptosis is critically regulated by mitochondrial function. Moreover, chondrocyte ATP depletion is driven via suppression of mitochondrial oxidative phosphorylation by nitric oxide (NO) as OA evolves in aging, thereby promoting increased ATP scavenging by NPP activity and consequent augmentation of extracellular PPi.74
Inflammation-associated chondrocyte hypertrophy is driven by hypoxia-inducible factor-2α and Indian hedgehog,60 as well as by multiple cytokines and calgranulins, oxidative stress, Pi transport, and receptor for advanced glycation end products (RAGE) signaling, and it is modulated by transglutaminase 2 (TG2) release. Chondrocyte hypertrophy and inflammation jointly drive chondrocalcinosis and progression of OA. For example, IL-1β, which is increased in OA cartilage, stimulates articular chondrocytes to calcify the matrix.51,64 NO stimulates both apoptosis and calcification in chondrocytes.71 IL-1β stimulates inducible nitric oxide synthase (iNOS) expression and increased NO generation, as well matrix alterations. IL-1β (as well as TNF, donors of NO, and the potent oxidant peroxynitrite) also induces increased chondrocyte transglutaminase (TG) activity mediated through the TG family enzymes, factor XIIIA and TG2.51,64
TG2 and factor XIIIA, which function in part to cross-link proteins by transamidation, are markers of growth plate chondrocyte hypertrophy.51,64 Significantly, there is upregulation of TG2 and factor XIIIA expression in hypertrophic cells in the superficial and deep zones of knee OA articular cartilage and the central (chondrocytic) zone of OA menisci.51 Moreover, increased factor XIIIA and TG2 activities both directly stimulate calcification by chondrocytes.51 OA severity–related, donor age–dependent, and marked age–dependent IL-1-induced increases in TG activity occur in chondrocytes from human knee menisci.51 TG2 is essential for IL-1β to stimulate articular chondrocytes to calcify in vitro.64 In addition, the closely related inflammatory chemokines CXCL1 and CXCL8, which are both increased in OA cartilage, induce TG275 and chondrocyte hypertrophic differentiation and calcification that requires TG2.75 Distinct TG2-independent and TG2-dependent mechanisms promote articular chondrocyte hypertrophy and calcification in vitro, and increased TG2 release is sufficient to promote chondrocyte hypertrophy.64,76 TG2 also promotes activation of TGF-β.77
The multiligand RAGE mediates several chronic degenerative diseases accompanied by low-grade inflammation.78 RAGE ligands include S100/calgranulins, a class of small, calcium-binding polypeptides, several of which are expressed by chondrocytes. Normal human knee cartilages demonstrate constitutive RAGE and S100A11 expression, and both RAGE and S100A11 expression are increased in OA cartilages. CXCL8 and TNF induce S100A11 release in cultured chondrocytes.78 Furthermore, S100A11 induces chondrocyte hypertrophy in vitro,78 and it does so in a manner dependent on S100A11 homodimerization catalyzed by TG2-mediated transamidation and antagonized by the alternative S100A11 receptor CD36.79,80 CXCL1-induced and TNF-induced chondrocyte hypertrophy require RAGE signaling.78
Special Pathogenic Aspects of Articular and Periarticular Basic Calcium Phosphate Crystal Deposition
CPPD and BCP crystal deposition can develop in different zones of articular cartilage and probably in distinct phases of cartilage degenerative disease such as ongoing loss of viability in hypertrophic chondrocytes. In addition, abundant cartilage NO production may promote mitochondrial dysfunction, chondrocyte extracellular ATP depletion,74 and lowering of extracellular PPi, consequently favoring HA over CPPD crystal deposition.81 The observation that OA and HA crystal deposition in articular cartilage (and arteries)30 are both promoted by extracellular PPi deficiency states strikingly illustrates the deleterious effects of deprivation of physiologic extracellular PPi levels.19 Yet it is noted that joint fluid PPi and NPP activity are elevated in HA-associated shoulder arthropathy (Milwaukee shoulder syndrome [MSS]),82 consistent with the model in Figure 96-1.
Pathologic BCP crystal deposition may occur in periarticular sites, as well as numerous organs and soft tissues. Significantly, the shoulder is the most common articular region affected by symptomatic BCP crystal deposition, in part reflecting unique shoulder structure-function (see Chapter 46). Degenerative changes promoted by biomechanical stress promote calcific tendinitis in the body of the rotator cuff.82 Such tendon calcifications can remain asymptomatic and also can eventually resorb, but the degenerative changes can predispose to tendon rupture. Osteopontin, a factor that normally restrains BCP crystal deposition (and is regulated by PPi and Pi),24 can be detected in fibroblast-like cells and multinucleated macrophages surrounding areas of calcification in calcific tendinitis.83 In this regard, osteopontin promotes oxidative stress, MMP activation, and macrophage recruitment and osteoclast activation.24 The presence of multinucleated cells with cathepsin K expression and osteoclast-like functions at sites of tendon calcification84 suggests a mechanism for both resorption of BCP crystal deposits and tendon degeneration.
Crystal-Induced Inflammation
Some of the crystals deposited in cartilage (Supplemental Figure 96-1 on www.expertconsult.com) can subclinically traffic to joint fluid and synovium, and the crystals can directly stimulate chondrocytes, synovial lining cells, and intra-articular leukocytes.85–91 Inflammation triggered by CPPD and BCP crystals thereby contributes to cartilage degradation and can potentiate worsening of OA.85–91 Many proinflammatory mechanisms active in gout also likely mediate synovitis and cartilage degeneration associated with CPPD and BCP crystal deposition.85–91 In this regard, CPPD and BCP crystals activate cells partly via nonspecific activation of signal transduction pathways (e.g., mitogen activated protein kinase activation) and induce cellular release of cyclooxygenase- and lipoxygenase-derived metabolites of arachidonic acid and cytokines including tumor necrosis factor (TNF), IL-1, and CXCL8.85–91 Innate immune recognition of extracellular CPPD crystals by Toll-like receptor 2 (TLR2)92 and CPPD crystal-induced activation of the intracellular NLRP3 (cryopyrin) inflammasome, resulting in caspase-1 activation and IL-1β processing and release, drive cell responses to CPPD crystals in vitro and CPPD crystal-induced inflammation in vivo.93
The ingress of neutrophils into the joint is central in triggering acute crystal-induced synovitis, and effects on neutrophil-endothelial interaction likely represent a major locus for prophylactic effects of nanomolar concentrations of colchicine for the acute arthritis of pseudogout.95 CXCL8 and related chemokines that bind the CXCL8 receptor CXCR2 (including CXCL1) appear to be critical in initiating and perpetuating neutrophil ingress in acute crystal-induced inflammation.96 Despite the fact that BCP and CPPD crystals share the capacity to activate certain cell signaling pathways and to induce several MMPs, BCP crystals generally trigger much less neutrophil influx into the joint than do CPPD crystals. Concordantly, free intra-articular BCP crystals likely induce less proinflammatory cytokine expression than do CPPD and monosodium urate crystals,97–99 though OCP crystals could be more inflammatory than hydroxyapatite crystals.100
Clinical Features
Calcium Pyrophosphate Dihydrate Deposition Disease
Most elderly individuals with CPPD deposition disease in the United States have a primary (idiopathic/sporadic) disorder (Table 96-2). Idiopathic chondrocalcinosis generally appears only after the fifth decade of life. But patients with a history of repetitive joint trauma of knee meniscectomy may present with nonsystemic (monoarticular) chondrocalcinosis before age 55. Familial forms of CPPD crystal deposition disease also have been widely documented, as discussed later. The clinical presentation of familial chondrocalcinosis is often manifested in the third and fourth decades of life, but familial disease can sometimes be detected before age 20 or first present clinically at advanced age. CPPD crystal deposition disease is also a common manifestation of a variety of hereditary and metabolic conditions (including hyperparathyroidism, dialysis-dependent renal failure, and hemochromatosis)55 in which the CPPD-related arthropathy can present earlier than age 55. For unclear reasons, hemochromatosis can present predominantly with CPPD crystal deposition disease or as OA. The weight of evidence from controlled studies suggests that hypothyroidism (with the probable exception of myxedematous hypothyroidism) is not associated with a significantly increased prevalence of CPPD crystal deposition disease, though both disorders are clearly prevalent in aging.55,101,102 It has been suggested that initiation of thyroxine supplementation therapy may trigger pseudogout.103
Table 96-2 Causes of Calcium Pyrophosphate Dihydrate Crystal Deposition Disease
| High Prevalence |
| Moderate Prevalence |
| Low Prevalence (Largely Based on Case Reports) |
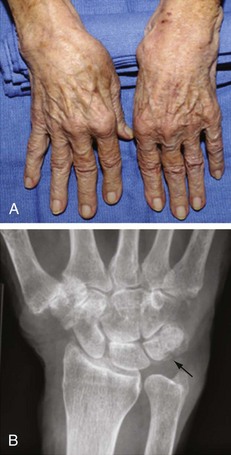
The clinical manifestations of CPPD deposition disease vary widely104 (Table 96-3). Quite commonly, the disease can be asymptomatic. Alternatively, it can mimic OA (pseudo-osteoarthritis), gout (pseudogout) (Supplemental Figure 96-2 on www.expertconsult.com), acute-onset or insidious rheumatoid arthritis (RA) (pseudorheumatoid arthritis) (Figure 96-4), or present as “pseudo-neuropathic” arthropathy. Patients with CPPD crystal deposition disease also commonly present with episodes of hemarthrosis, often post-traumatic and in the knee. The contributions of the forms of CPPD deposited (e.g., monoclinic vs. triclinic crystals) and of host factors to these wide differences in clinical manifestations are not clear. Overall, only a small fraction of patients with CPPD deposition disease have prolonged, recurring polyarticular inflammation. Progressive degenerative arthropathy is more common. Though CPPD deposition disease appears to be a common and significant public health problem in the elderly, the disease and health-related quality of life impact and the long-term course of CPPD-associated degenerative arthropathy in an unselected population have not been adequately evaluated.
Table 96-3 Common Clinical Presentations of Calcium Pyrophosphate Dihydrate (CPPD) Crystal Deposition Disease
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


