20 Autoimmunity
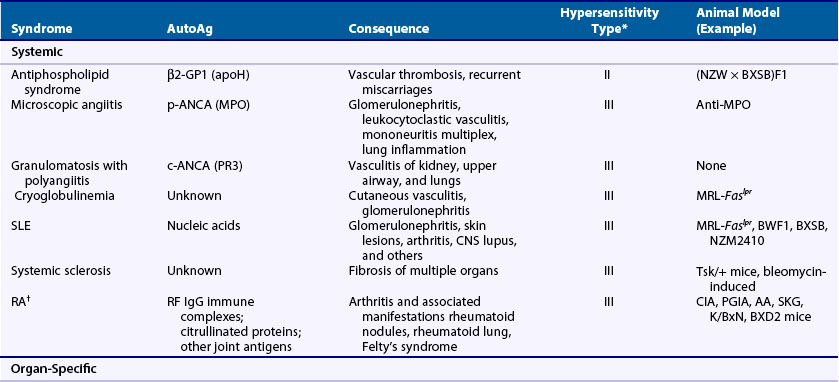
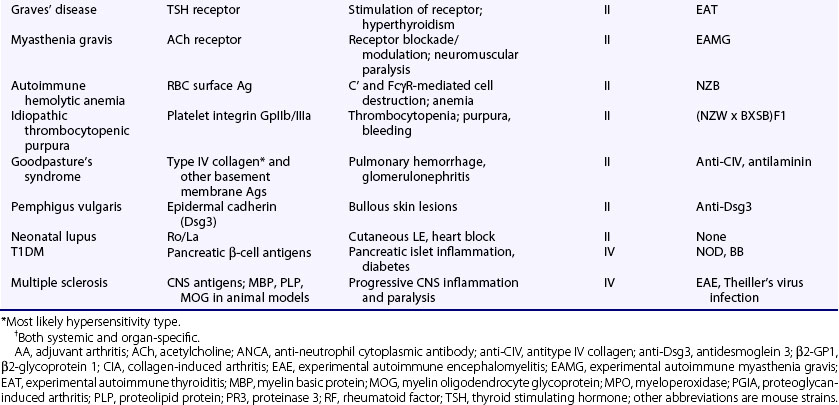
Autoimmune diseases represent a significant health burden for 3% to 9% of the general population, and rheumatology, perhaps more than any medical subspecialty, encompasses a broad array of such diseases involving a wide range of organ systems (Table 20-1).1–3 Rheumatologists consequently have a substantial interest in defining the causes and pathophysiologic processes related to autoimmunity and in applying this information to the clinic.
Table 20-1 Examples of Rheumatologic Autoimmune Diseases
CREST, calcinosis, Raynaud’s phenomenon, esophageal dysfunction, sclerodactyly, and telangiectasia.
Definition and Classification of Pathogenic Autoimmunity
Autoimmunity, the immune response against self, evokes the specter of “horror autotoxicus,” a term coined by Paul Ehrlich at the turn of the 20th century to depict the perceived disastrous consequences of this condition.4 In fact, autoreactivity is more nuanced, ranging from a low “physiologic” level of self-reactivity that plays an essential role in lymphocyte selection and maintenance of normal immune system homeostasis, to an intermediate level of autoimmunity including autoantibodies and tissue infiltrates unassociated with clinical consequences, to pathogenic autoimmunity associated with immune-mediated dysfunction or injury. From the clinical perspective, it is the transformation to pathogenic autoimmunity that demarcates significant from insignificant self-reactivity.
An example of diseases that exhibit some characteristics of autoimmunity yet are distinct in their pathogenesis are the so-called autoinflammatory syndromes.5–7 These are mostly rare monogenic disorders typified by intermittent bouts of fevers, rash, serositis, and arthritis caused by defective control of basic inflammatory mechanisms. Included are familial Mediterranean fever, the cryopyrinopathies, hyperimmunoglobulinemia D with recurrent fever, familial cold urticaria, and Blau syndrome. These disorders could be considered one end of a broader definition of autoimmunity, but because their pathophysiology is mediated entirely through the innate arm of the immune system, they are currently classified as a separate entity. Nonetheless, the possibility of a less stringent demarcation has been suggested by disorders such as Behçet’s syndrome, systemic juvenile rheumatoid arthritis, and Crohn’s disease, which appear to manifest both autoinflammatory and autoimmune features.
Autoimmune diseases are classified as systemic or organ-specific depending on the extent of their clinicopathology (Table 20-2). The systemic category includes systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), scleroderma, primary Sjögren’s syndrome, dermatomyositis, and systemic vasculitis. In this case, autoimmunity targets ubiquitously expressed self-antigens and end-organ injury is typically mediated by autoantibodies and, less commonly, T cells. Contrastingly, in organ-specific diseases the self-antigens are typically cell or tissue specific in location or accessibility and end-organ damage can be mediated by antibodies and/or T cells. Some of the more notable examples in this group, which span virtually all organ systems, include Hashimoto’s thyroiditis, Graves’ disease, multiple sclerosis (MS), type 1 diabetes mellitus (T1DM), antiphospholipid syndrome (APS), pemphigus vulgaris, autoimmune hemolytic anemia, idiopathic thrombocytopenic purpura, and myasthenia gravis. It should be noted, however, that although the distinction of systemic and organ-specific disorders provides a conceptual framework, the pathophysiologies of autoimmune diseases are more diverse than might be implied by this simple classification. Autoimmune diseases can also be classified by hypersensitivity reaction type based on the mechanism of adaptive immune system–mediated injury.8 See later discussion in the chapter.
Animal Models of Autoimmunity
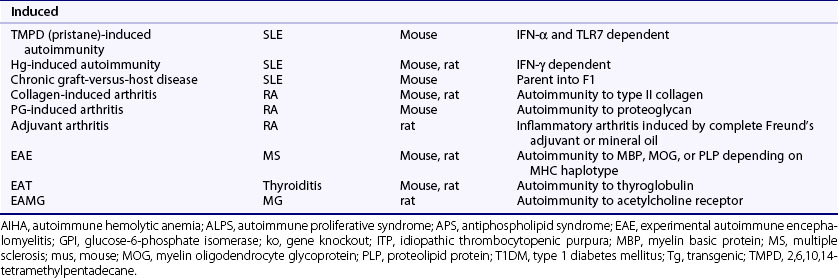
Much of what is known about the immune system and autoimmunity has been derived from studies in animals, particularly the mouse, which has an immune system and genome composition similar to humans. There are many well-characterized autoimmune models, and investigators can manipulate their genomes and immune systems, test interventions, and modify their environments. Animal models of autoimmune disease comprise three main types on the basis of their derivation: (1) spontaneous, (2) genetically modified, and (3) induced. Table 20-3 lists some of the more common or notable types (see also Chapter 28).
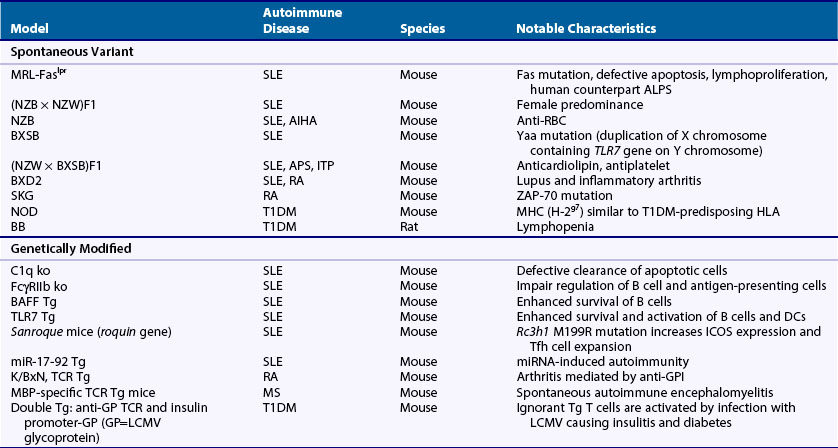
Within the spontaneous group are models of SLE, RA, and type 1 diabetes mellitus. The lupus-prone strains commonly develop anti-DNA and immune-complex-mediated kidney damage, but they also possess unique phenotypic characteristics and differ in the genes underpinning susceptibility.9 The SKG arthritis model is a spontaneous, inflammatory, and erosive arthritis caused by a ZAP-70 mutation that, similar to RA, is associated with rheumatoid factor (RF) and antibodies to citrullinated proteins.10 The NOD mice and BB rats develop T1DM caused by T cell–mediated destruction of β-islet cells.11
The genetically modified group, encompassing transgenic, site-directed genetic replacement (gene knockout or knockin), and ENU-mutagenized mice, is by far the largest, with more than 80 different models of lupus alone, as well as many models of organ-specific diseases, particularly T1DM and MS.9,11 The lupus models, mostly single-gene knockout or transgenic mice, have provided a wealth of information related to immune tolerance and disease pathogenesis. Some examples are (1) confirmation of human SLE gene associations and elucidation of mechanisms (e.g., C1q and FcγRIIb knockout mice), (2) discovery of novel mechanisms (e.g., altered mRNA regulation in Sanroque and miR-17-92 transgenic mice), and (3) identification of new pathways relevant to therapy (e.g., BAFF transgenic). An example of an arthritis model created by genetic modification is the K/BxN mouse, a B6 × NOD hybrid expressing a transgenic T cell receptor, named KRN, that recognizes a bovine RNase peptide on MHC class II Ak.12 These mice develop an acute severe inflammatory arthritis caused by antibodies to glucose-6-phosphate isomerase (GPI), an antigen that, although intracellularly and ubiquitously expressed, is mainly accessible to antibodies in joints. GPI is not a major target in RA or other arthritides, but the model has nevertheless been useful for investigating inflammatory mechanisms in arthritis.13 Other genetically modified models of spontaneous arthritis include the HTLV-I tax transgenic, the TNF transgenic, the IL-1ra transgenic, the IL-1 transgenic, and a gain-of-function knockin mutant of CD130, a signaling component for several cytokines including IL-6, IL-11, IL-27, and LIF.14 Scientists have developed several genetically modified models of T cell–mediated organ-specific disease. They consist of TCR transgenic T cells that recognize tissue-specific antigens in organs such as the brain and pancreatic islets or employ a slightly modified version in which mice are double transgenic for both an antigen expressed in a tissue of interest and the corresponding TCR.15–18 By allowing analysis of single autoreactive T cell clones, these models have yielded considerable insights into tolerance mechanisms and disease pathophysiology. Similarly, researchers have developed autoreactive B cell receptor transgenics or knockin models that have been crucial for defining B cell tolerance mechanisms.19–25
The induced models encompass a wide variety of both systemic and organ-specific diseases. More commonly studied models of systemic disease include tetramethylpentadecane (TMPD, also called pristane)-induced autoimmunity, mercury-induced autoimmunity, and chronic graft-versus-host disease.26–28 All bear similarities to human SLE in producing antinuclear antibodies and immune complex deposits, but they differ in pathophysiology and strain susceptibility. For the induced models of organ-specific diseases, a common approach is to immunize rodents with a self-antigen or closely related peptide or foreign-antigen, plus a strong adjuvant, usually complete Freund’s. This approach makes it possible to induce autoimmunity in virtually all organ systems and to produce diseases mediated by cellular and humoral mechanisms. Some of the more commonly studied organ-specific models developed by this approach include collagen-induced arthritis (CIA), proteoglycan-induced arthritis (PGIA), and experimental autoimmune encephalomyelitis (EAE), but there are many others to thyroid, eye, gonad, nerves, neuromuscular junction (acetylcholine receptor), muscle, heart, adrenal gland, bladder, stomach, liver, inner ear, kidney, and prostate tissues. In certain susceptible strains of rats, a progressive inflammatory erosive arthritis called adjuvant arthritis can also be induced by intradermal injection of complete Freund’s adjuvant or by mineral oil components of Freund’s adjuvant such as TMPD. Other induced models of arthritis include streptococcal cell wall and antigen-induced arthritis.14 Recently, arthritis characterized by synovial hyperplasia and ankylosis was reported in HLA-DR4 transgenic mice immunized with human citrullinated fibrinogen.29
Composition of the Innate and Adaptive Immune Systems
The immune system comprises two major arms, innate and adaptive. The former is classically the initiator of immune responses to pathogens because of its capacity, on the one hand, to be activated by diverse pathogen-associated molecular patterns (PAMPs) expressed by microbes and, on the other hand, to activate the adaptive immune system through the production of proinflammatory factors and the presentation of antigens to lymphocytes.30–32 The adaptive response adds more protection through clonal recognition of an almost limitless diversity of antigenic specificities, immunologic memory of previous antigenic encounters, and additional cellular and antibody-mediated mechanisms to eliminate or neutralize microbes and other pathogens. Both systems use a large variety of immune-related molecules, cell types, and interactions among the various components to provide layers of protection that contribute to overall resistance to commensal and invading organisms. Similarly, prevention of self-reactivity is also effected by multiple inhibitory or deleting processes that, in toto, afford layers of safeguards against autoimmunity. The innate and adaptive immune systems are highly interconnected with activating and inhibitory feedback mechanisms operating in both directions.
Innate Immune System Composition
The main functions of the innate immune system include (1) the clearance of foreign and potentially detrimental self-materials through the recruitment, activation, and effector activity of immune cells; (2) activation of the complement pathways, and (3) triggering of the adaptive immune response (see also Chapter 18). A key feature of the innate, and to some extent the adaptive, immune system is that it is activated by the engagement of pattern-recognition receptors (PRRs) that recognize common structures on pathogens and also a few self-molecules such as certain damage-associated molecular patterns (DAMPs) (e.g., heat shock proteins),33 released during cellular stress, injury, or death. PRRs can be classified by their location into secreted, transmembrane, or cytosolic types.31 Secreted PRRs include collectins, pentraxins, ficolins, and to some extent C1q, properdin, serum amyloid A, and mindin, which facilitate clearance of pathogens by promoting opsonization and complement activation.34 Transmembrane PRRs that provide activating signals following engagement with the corresponding PAMPs include the Toll-like receptors (TLRs), several C-type lectin receptors (dectin receptors, mannose receptor, DC-SIGN), and N-formyl methionine receptors.35
The TLRs are of particular interest because of their role in autoimmune diseases such as SLE.36 They are a family of type I membrane proteins that contain a ligand-binding ectodomain composed of 18 to 25 tandem leucine-rich repeats, a transmembrane region, and an intracellular cytoplasmic Toll/interleukin 1 receptor (TIR) domain through which they interact with two main adapters, MyD88 (all TLRs except TLR3) and TRIF (TLR3, TLR4).37,38 TLRs are either expressed as cell surface receptors where they recognize primarily bacterial products such as lipoteichoic acids (TLR1/2 heterodimer), lipoproteins (TLR2/6), lipopolysaccharide (TLR4), and flagellin (TLR5), or in the endolysosomal compartment where they bind different types of nucleic acids including dsRNA (TLR3), ssRNA (TLR7), and dsDNA (TLR9). The intracellular location of the nucleic acid–binding TLRs is thought to reflect both the optimal location for exposure to microbial nucleic acids, which are released in the endolysosomes following phagocytosis, and the necessity of avoiding activation by extracellular self-nucleic acids.31 Cell types express different combinations of TLR members, reflecting the role of specific TLRs in cell function (e.g., the endosomal TLRs recognizing nucleic acids are predominantly expressed in phagocytes and antigen-presenting cells).
The cytosolic PRRs include (1) RIG-I-like receptors (RLRs) that include RIG-I, a sensor for foreign uncapped 5′-triphosphate RNA and MDA5 that recognizes long dsRNA39,40; (2) NOD-like receptors (NLRs) that recognize bacterial cell wall products41,42; and (3) several DNA sensors, IFI16,43,44 LRRFIP1,45 Ku70,46 AIM2, and DNA-dependent RNA polymerase III, which does not detect DNA directly but transcribes AT-rich DNA to 5′uncapped 5′-triphosphate RNA that in turn activates RIG-I.7,32,47,48 The role of these cytosolic PRRs in autoimmune diseases has not yet been defined.49
Collectively, engagement of the TLR and cytosolic PRRs activates immune cells through several signaling pathways that converge primarily to activate nuclear factor κB (NFκB)/cytokine or inflammasome programs. Furthermore, depending on the PRRs engaged, cell types activated, and site of activation, this system of receptors facilitates the generation of nuanced responses to a wide array of potential pathogens.50
Major innate immune system cell types include macrophages, dendritic cells, neutrophils, plasmacytoid dendritic cells (pDC), natural killer (NK) cells, basophils, mast cells, and eosinophils. In addition, certain T lymphocyte subsets that express predominantly invariant antigen receptors that recognize microbial products such as certain γδ-T cells51 and NK T cells52 might also be considered part of the innate system. Similarly, certain B lymphocyte subsets, including B1 and marginal zone B cells, have characteristics suggesting a lineage with innate properties (e.g., development from precursors early, but not late in life, the capacity for self-renewal and expression of primarily low-affinity polyreactive IgM).
The innate response is also strongly influenced by nonhematopoietic cells including endothelial, stromal, fibroblast, and keratin cells that can elaborate proinflammatory factors following engagement of PRRs, injury, or activation by the adaptive immune system. Examples include, nonhematopoietic cells are required for NOD 1 receptor-mediated Th2 immune responses53, synovial fibroblasts play a central role in RA54, and keratinocyte secretion of CSF-1 after UV exposure promotes cutaneous lupus.55
The complement system56 comprises an immune surveillance system that interacts with, and promotes elimination of, modified host cells, cellular debris, and foreign entities (see also Chapter 23). This system is tightly interconnected with inflammatory, immune, and other pathways and because of this, often plays important roles in both autoinflammatory and autoimmune diseases.
Adaptive Immune System Composition
The main constituents that distinguish the adaptive from the innate immune response are the B and T lymphocytes, which express an expansive array of diverse antigen receptors (see also Chapter 19). In aggregate, the pool of lymphocytes is capable of recognizing virtually all antigens and, by the process of selection, lymphocytes with beneficial specificities can be expanded and retained. These properties allow the immune system to detect a manifold galaxy of foreign antigens and to respond rapidly and specifically to secondary challenges. T and B lymphocyte antigen receptors are created by the random recombination of a large number of variable region (V-D-J) gene segments and diversity-producing end-joining mechanisms in primary lymphoid organs. Furthermore, peripheral B cells can also increase specificity by somatic hypermutation and selection of higher affinity clones. The clonal nature of lymphocytes, bestowed by uniquely rearranged antigen receptors, is key to the exquisite discriminating power of the immune system. Clonality also extends to other characteristics such as cytokine production, surface receptors, and cell signaling molecules.
T cells play a central role in the adaptive immune response through elaboration of cytokines such as IFN-γ and TNF, IL-4, and IL-17, by providing essential signals to B cells in T cell–dependent humoral responses, as direct killers of cells, and by promoting inflammation in target tissues (see also Chapter 13). Importantly, because of their clonal nature and the immune system’s ability to neutralize specific self-reactive clones, they play a fundamental role in regulating autoreactivity. T cells arise in the thymus, where they transit through defined maturation stages and undergo positive and negative clonal selection.57,58 Thymic-derived T cell populations include the classical CD8 and CD4 αβ T cells, γδ T cells, NK T cells, natural regulatory CD4 T cells (nTreg), and a subset of NK cells.59 Recent thymic CD4 and CD8 T cell emigrants mature in the periphery, where, unless activated, they can remain as circulating naïve T cells for the lifespan of the organism. Following activation by antigen-presenting cells, T cells expand and differentiate into several possible subsets. For CD4 T cells, these include the major Th1, Th2, Th17, follicular helper (Tfh), induced Treg (iTreg) subsets, and the less well-defined Th9 and Th22 populations that promote or inhibit distinct immune response activities.60–62
B cells develop in the bone marrow, where, like T cells, they undergo defined maturation stages and selection of antigen receptors (see also Chapter 14). In addition, class-switch recombination and somatic hypermutation of the B cell receptor occurs during T cell–dependent humoral responses. The main B cell types in the periphery include the conventional (or B2) and B1 subsets, with B2 cells further divided into follicular and marginal zone subsets.
Tolerance Mechanisms
Increasing insights into mechanisms of self-nonself discrimination have emerged over the past 6 decades in parallel with growing knowledge of the immune system. More than 50 years ago, Burnet and Medawar advanced the critical concept that tolerance was imposed by clonal deletion of self-reactive lymphocytes during early development (i.e., central tolerance).63,64 Later, with the discovery that mature B cells undergo somatic hypermutation in the periphery, it was hypothesized by Bretscher and Cohn that the production of autoantibodies might be impeded by the need for both B and T cell compartments to breach tolerance.65 In 1975, while studying allogeneic responses, Lafferty and Cunningham posited that activation of T cells involved the passing of a second signal that need not be antigen-related, thereby implicating costimulation from antigen-presenting cells as a critical factor in lymphocyte activation.66 In 1987, the nature of the costimulation, or two-signal, requirement was further defined when Jenkins and Schwartz showed that engagement of antigen receptor alone without a second signal resulted in functional inactivation of T cells.67 A novel mechanism for self-nonself discrimination that incorporated the innate immune system was then advanced by Janeway in 1989 when he hypothesized that antigen-presenting cells critical for T cell activation remain quiescent unless activated by the engagement of PRRs by microbial products.68 This concept was further extended by Matzinger in 1994 to a “danger model” that includes activation of the immune system by both foreign and endogenous factors associated with tissue stress and damage.69 These models have laid the foundation for the current, more complex, view of self-nonself discrimination in which tolerance is imposed by both innate and adaptive immune systems through layers of mechanisms occurring at various stages throughout lymphocyte development and activation (Table 20-4).
Clone-Specific Self-Nonself Recognition
In contrast to innate immune cells, which are activated primarily by hardwired microbial PRRs, lymphocytes are unrestricted in specificity and therefore self-nonself discrimination must be implemented at the clonal level. To achieve this, T and B cells use several mechanisms that can be grouped into three general strategies. First, the type of response is controlled by developmental stage. For example, immature lymphocytes respond to strong antigen receptor stimulation by cell death, whereas a similar signal in mature cells leads to activation. Through this mechanism self-reactive clones are eliminated from the nascent lymphocyte repertoire before they can cause injury. Second, activation of mature lymphocytes requires—in addition to antigen receptor engagement—a second co-stmulatory signal that, if absent, results in anergy or cell death. For the most part, this requirement limits reactivity to self because co-stimulatory signals are largely provided by activated cells of the innate immune system. Third, lymphocytes are fine-tuned in various ways by a fairly extensive list of modulating factors, which is necessary for controlling self-reactive clones.70–72 For example, defects affecting a broad range of surface receptors on lymphocytes, including those with prosurvival (IL-7R, BAFFR, IL-2R); proapoptotic (TNFRs, FasL, TRAIL); costimulatory (CD28, CD40, TLRs); differentiating (IL-12R, IL-4R, IFNγR, IL-23R, retinoic acid R, transforming growth factor (TGF)-βRs, SAP/SLAM family members, OX40, ICOS/ICOSL); inhibitory (FcγRIIb, CD22, CTLA4, PD-1); antigen receptor signal modulating (CD19, CD45); and activating (SAP/SLAM family members) activities have been shown to influence the development of autoimmunity.9 Collectively, these self-nonself recognition mechanisms provide the basic cellular means by which the innate and adaptive immune systems render T and B cell clonotypes tolerant to self-antigens and resistant to autoimmune disease.
Innate System and Tolerance
Given its vital role in initiating and modulating adaptive immune responses, it is not surprising that the innate immune system strongly influences both tolerance and autoimmunity. Indeed, although its contributions have yet to be fully explicated, several ways in which self-tolerance is influenced by the innate arm have been defined. First, as noted previously, activation of the innate immune system under normal circumstances typically requires engagement of microbial PRRs, which endows the immune system with a direct and simple way to distinguish foreign- from self-antigens.73 The importance of this mechanism is illustrated by the finding that overexpression of TLR7 by spontaneous gene duplication or transgenic approaches promotes systemic autoimmunity.36 This occurs because certain PRRs, such as TLR7 and TLR9 that equally sense both foreign and self-nucleic acids, avoid significant exposure to endogenous nucleic acids by virtue of their location in subcellular compartments.74 However, in the case of TLR7, overexpression makes it possible for normally substimulatory amounts of endogenous RNA to activate immune cells, thereby bypassing the usual requirement for microbial exposure. Second, some cells of the innate immune system actively suppress adaptive immune system activation under certain conditions. For example, immature and, under some circumstances, even mature dendritic cells have been shown to promote tolerance by inducing CD4 T regulatory (Treg) cells and other mechanisms.75 Third, another critical function of the innate immune system is the rapid noninflammatory clearance of apoptotic cells.76 Failure can result in an increased supply of self-antigenic material including nucleic acids, secondary necrosis, and release of proinflammatory factors, leading to systemic autoimmunity.77 Accordingly, deficiencies in several key apoptotic cell clearance molecules are associated with autoimmunity including (1) the Tyro3-Axl-Mer receptors on phagocytes that bind through Gas6 or protein S, the exposed phosphatidylserine (PS) on apoptotic cells78; (2) the milk fat globule-EGF factor 8 (MFG-E8) protein that bridges the αvβ3 integrin on phagocytes and PS on apoptotic cells79,80; and (3) natural IgM or C1q that binds to and enhances clearance of apoptotic cells.81–83 Fourth, several complement components have been directly implicated in autoimmunity. For example, SLE is associated with deficiencies of proximal components of the classical pathway including C1q, C4, and C2. The mechanism for this is not certain, but both defective clearance of apoptotic material/immune complexes and a shift in the activation threshold of lymphocytes have been suggested.84 As another example, deficiency of CD55 (or decay-accelerating factor), a cell surface protein that restricts complement activation, is associated with enhanced T cell responses and exacerbation of neuroinflammation and lupus in animal models.85,86 Thus at many levels, the innate immune system plays a critical role in maintaining tolerance and controlling autoimmunity.
T Cell Tolerance
T cells are critical players in both achieving and fine-tuning tolerance to a high degree of specificity. Researchers have identified several mechanisms and divided them into three main areas: central tolerance wherein T cells first acquire their antigen receptor, peripheral tolerance wherein T cells encounter self-antigens not present in the thymus, and postactivation regulation wherein activated and expanded T cell clones are returned to their resting state. Central tolerance, as alluded to earlier, is imposed on T cells with self-reactive specificities during thymocyte development by mechanisms using primarily deletion, to a lesser extent anergy, and possibly receptor editing of the TCR α-chain.87,88 This process, although highly effective, is not completely efficient, and T cells with autoreactivity, primarily those with intermediate to low affinity to self or recognizing self-antigens poorly expressed in the thymus, emigrate in significant numbers. This leakiness is probably necessary to generate a broad repertoire, but it then creates vulnerability to autoimmunity and necessitates peripheral tolerance mechanisms.
In the periphery, multiple mechanisms for avoiding autoreactivity have been identified. Among these, a common explanation is that most tissue-associated self-antigens are not accessible to trigger a response, a situation that can be caused by low abundance, specific characteristics of the self-antigen, or location. This mechanism is supported experimentally by the finding that T cells expressing a transgenic TCR to certain tissue-specific antigens are not deleted or activated, nor do they cause autoimmune disease. Yet these so-called “ignorant” T cells are fully functional and respond to self-antigen when presented in a conventional context such as inflammation or tissue damage.18,89 For a few self-antigens such as those expressed in the anterior chamber of the eye, central nervous system, or other so-called immunologic privileged sites—as originally defined by their ability to accept allograft transplants—resistance to self-reactivity also partly arises from anatomic sequestration caused by limited access of blood-borne cells and the absence of conventional lymphatic drainage.90 The latter is important because T cells are typically first activated in secondary lymphoid organs and subsequently migrate to target organs, where reactivation by local antigen-presenting cells and the production of proinflammatory factors leads to tissue damage.91 Anatomic sequestration alone, however, is not sufficient for tissues to support immunologic privilege and, as discussed later, such sites employ a host of additional local mechanisms.92,93
Another peripheral mechanism is the aforementioned two-signal paradigm wherein T cell activation requires both TCR engagement and a co-stimulatory signal usually provided by CD28. Because the two ligands for CD28, CD80 and CD86, are primarily expressed at high levels on activated professional antigen-presenting cells, presentation of self-antigen by quiescent antigen-presenting cells would lead to tolerance. Indeed, immature DCs promote tolerance in this manner by constitutively presenting low doses of self-antigen on MHC, resulting in cell death or anergy of the corresponding T cells.94
Peripheral tolerance of T cells is also maintained by active suppression via immunoregulatory cells of the immune system, among which CD4+ Tregs are the best characterized. They constitute a distinct αβ T cell subset generated in the thymus (natural Treg, nTreg) or in the periphery from naïve or mature CD4+ T cells exposed to TGF-β (induced Treg, iTreg). Both are developmentally induced by the Foxp3 (forkhead boxP3) transcription factor. They typically express high levels of the IL-2 receptor component, IL-2Rα (CD25), and require IL-2 for survival. Tregs participate in every adaptive immune response, are critical for maintaining the proper level of immune response, and are activated at the same time as conventional T cells. They are thought to suppress the magnitude of the response by (1) initially downregulating DC function and then inhibiting T cell activation by competing for IL-2, (2) producing immunosuppressive cytokines such as TGF-β, IL-10, and IL-35, and (3) by cell-cell inhibitory interactions that lead to cell killing or the induction of negative signals.95 These inhibitory actions suppress T cells that are in proximity to Treg cells regardless of their antigen specificity.96 Researchers have described other T cells with regulatory activity in autoimmunity including Tr1, CD8 Treg, Qa-1/HLA-E-restricted CD8 T cells, and γδ T cells, but they are less well characterized.97–101
Tissues themselves also employ mechanisms that suppress self-reactivity and contribute to establishing immune privilege.92,102–104 These comprise three general categories. First, certain tissues are decorated with cell surface inhibitory molecules such as the proapoptotic FasL and TRAIL, lymphocyte inhibitory and Treg-promoting PD-L2, and complement regulatory proteins, CD55 and CD46, that can potentially eliminate or impede the activation of autoreactive T cells. Second, soluble inhibitors of inflammation and immune activation are secreted by particular tissues. Notably, in the aqueous humor of the eye, there is a broad spectrum of such factors that include TGF-β, α-melanocyte-stimulating hormone, vasointestinal peptide, calcitonin gene–related peptide, somatostatin, macrophage inhibitory factor, and complement inhibitors. Third, lymph node resident stromal cells have been shown to induce tolerance of CD8+ T cells recognizing peripheral tissue-restricted self-antigens.105,106 Thus, it has been proposed that stromal cells in lymph node and tissues may provide a means to eliminate T cells that bind to tissue-restricted antigens not expressed in the thymus. Fourth, the anterior chamber of the eye elicits a unique type of altered immune response through a complex multistep process termed anterior chamber-associated immune deviation (ACAID) that leads to a dampened and less tissue-destructive response.92,104,107 Although ACAID was long thought important for tolerance, it was recently argued that its primary function may be to modulate the immune response so that the eye can respond to infection without damaging its integrity.108
Another possible peripheral mechanism is immune deviation wherein polarization away from a predisposing cytokine pattern, such as from a Th1 to a Th2 profile, suppresses the development of autoimmune disease.109 Similarly, activation of NKT cells with α-GalCer, which induces IFN-γ production, is associated with dampening of the adaptive Th1 and Th17 effector responses and protection from experimental autoimmune uveitis.110 In these models, autoreactive T cells are activated but do not produce the proinflammatory factors necessary for tissue damage.
B Cell Tolerance
B cells are required not only for antibody production but also serve as potent antigen-presenting cells for T cells and follicular DCs, and can act in regulation as well.111 Moreover, depletion of B cells with rituximab has shown promise even in autoimmune diseases considered to be mediated by T cells such as T1DM112 and multiple sclerosis.113 Therefore, there is substantial interest in defining both mechanisms of B cell tolerance and the specific role of B cell tolerance in autoimmune disease.
Central tolerance of B cells takes place in the bone marrow during preB to immature B cell transition as they express rearranged immunoglobulin (Ig) genes on their surface.114 It appears that the dominant mechanism for B cells with high affinity to membrane-bound self-antigen is receptor editing (replacement of L-chain) and, to a lesser extent, deletion, while soluble self-antigens induce both receptor editing and anergy.115,116 Anergic B cells are detectable in the periphery as an IgD+IgM− population117 or in mice as splenic transitional 3 (T3) B cells.118 They are short-lived at least in part because they downregulate the BAFF receptor, which is required for their survival, putting them at a competitive disadvantage with other immature B cells, and they are less able to enter B cell follicles.
In the periphery, the earliest tolerance checkpoint occurs at the transitional 1 (T1) B stage over a 2-day interval before maturation to T2 and later naïve B cell subsets.114,119–121 T1 B cells are the immediate bone marrow emigrant population, retain an immature phenotype, and are BAFF-dependent for survival. Importantly, they undergo apoptosis and not activation when stimulated, which results in deletion of B cells recognizing peripheral self-antigens not expressed in the bone marrow compartment. Thus, this mechanism, which is unique to B cells, represents in essence an extension of central tolerance to the periphery.
Other peripheral tolerance mechanisms are achieved through many of those described earlier for T cells but differ qualitatively due to distinct differentiation pathways and differences in antigen recognition by B cells and T cells (i.e., BCRs can bind to virtually all tertiary structures while TCRs are restricted to recognizing self-MHC/peptide complexes on host cell surfaces). Accordingly, B cells can be ignorant of their corresponding self-antigen because of insufficient quantity or access19 or can undergo anergy and ultimately cell death if there is engagement of the BCR without co-stimulation (i.e., two signals).122
Another notable checkpoint occurs during T cell–dependent immune responses as B cells undergo affinity maturation in germinal centers (GCs) and acquire new specificities, which may include self-reactivity. Evidence suggests that tolerance at this juncture is often defective in autoimmune diseases because most autoantibodies have acquired autoreactivity through somatic hypermutation and are class-switched,123–125 both indicative of GC maturation. Although recent studies have provided significant insights into GC processes involved in the selection of B cell clones with high affinity to foreign antigens, how tolerance of class-switched autoreactive B cells is achieved remains to be clarified. Nevertheless, the strongest evidence suggests (1) autoreactive B cells compete poorly for the cognate T cell help essential for GC B cell survival because the autoreactive BCR would bind less well to the original antigen, resulting in less internalization of antigen for processing and presentation to T cells126–128 and (2) B cells that acquire BCRs with high affinity to membrane antigens are deleted by a Fas-dependent mechanism.129
Theories of Autoimmunity
Development of autoimmune diseases is influenced by genetic and, to varying degrees, environmental, gender, and other factors, with current evidence supporting a model in which genetic predisposition is required (Figure 20-1). Therefore, theories of autoimmunity and loss of tolerance are closely intertwined with genetic influences. In addition, such theories must also explain how tolerance is breached when autoimmunity is induced in otherwise normal animals. Taking both these factors into account and applying a reductionist perspective, theories of autoimmune disease can be consolidated into two main mechanisms representing separate ends of a continuum—most diseases having some elements of both. On one end, loss of tolerance and the consequent autoimmune disease is caused by genetically imposed defects in central and/or peripheral tolerance mechanisms while, on the other end, autoimmunity arises from the conventional immune response to self-antigens for which tolerance is normally incompletely established (Table 20-5). In general, most systemic autoimmune diseases are caused by tolerance defects, whereas organ-specific diseases can be mediated by either mechanism.
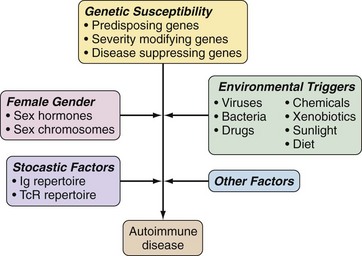
Table 20-5 Mechanisms of Autoimmunity
| Examples | Disease | Mechanism |
|---|---|---|
| Defective Tolerance | ||
| Central Defects | ||
| AIRE deficiency | APECED syndrome | Failure to delete autoreactive T cells because of reduced expression of peripheral antigens in thymus |
| ZAP-70 deficiency | Inflammatory erosive arthritis (mice) | Defective T cell activation and thymic selection |
| Peripheral Defects | ||
| FAS/FASLG deficiency | Autoimmune lymphoproliferative syndrome (ALPS) | Defective apoptosis |
| Rc3h1 (M199R) mutation | Lupus (mice) | Increased ICOS on Tfh cells promotes their expansion |
| TREX1 (DNase III) deficiency | Aicardi-Goutières syndrome, chilblain lupus | Accumulation of intracellular DNA induces IFN-α production |
| FOXP3 deficiency | IPEX syndrome | Absence of Treg cells |
| PD-1 deficiency | Lupus, myocarditis (mice) | Defective peripheral tolerance of T cells |
| Activation of Nontolerant Lymphocytes | ||
| Penetrating injury | Sympathetic ophthalmia | Release of self-antigen in an inflammatory milieu |
| Coxsackie B virus infection | T1DM (mice) | Infection-mediated release of self-antigen in an inflammatory milieu |
| Immunization with self-antigen and strong adjuvant | EAE (mice) | Activation of ignorant T cells |
| Citrullination of proteins | RA | Generation of neo self-antigens |
| Altered structure of collagen IV caused by sulfilimine bonds | Goodpasture’s syndrome | Formation of conformational neo self-antigens |
| Cross-reactivity of group A streptococcal and cardiac antigens | Rheumatic fever | Molecular mimicry |
| Lymphopenia caused by disease associated IL-21 production | T1DM (NOD mice) | Lymphopenia-induced homeostatic proliferation |
AIRE, autoimmune regulator; APECED, autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy; EAE, experimental autoimmune encephalomyelitis; FASLG, FAS ligand; FOXP3, forkhead box P3; ICOS, inducible T-cell co-stimulator; IFN-α, interferon-α; IL-21, interleukin-21; IPEX, immune dysregulation, polyendocrinopathy, enteropathy X-linked; NOD, non-obese diabetic (mouse strain); PD-1, programmed cell death 1; RA, rheumatoid arthritis; T1DM, type 1 diabetes mellitus; TREX1, three prime repair exonuclease 1; ZAP-70, zeta-chain-associated protein kinase 70.
Defective Tolerance
Defects in central tolerance have long been suspected to cause autoimmunity because of its well-documented role in eliminating nascent self-reactive lymphocytes, but until recently solid evidence for this was lacking. A breakthrough came from the discovery that mutations in a transcription factor, AIRE (for autoimmune regulator), caused autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), a rare inherited disease associated with T cell and autoantibody–mediated autoimmune destruction of multiple endocrine organs.130,131 AIRE-deficient mice developed a similar syndrome, which was caused by reduced expression of thousands of peripheral tissue genes in the thymic medullary epithelium and consequently failure to eliminate T cells specific for those gene products. Thus it appears that the main function of AIRE is to prevent autoimmunity by deleting T cells recognizing peripheral tissue self-antigens.
Another example of altered thymic selection leading to autoimmunity, but in this instance caused by a defect in T cells, is the aforementioned SKG arthritis model. Here, a function-impairing mutation in the C-terminal SH2 domain of ZAP-70, a Syk family tyrosine kinase activated by the T cell receptor complex ζ-chain, reduces TCR signaling, leading to defective conventional T cell and nTreg development in the thymus, and presumably enhanced positive selection of autoreactive T cells.132 Interestingly, using several ZAP-70 mutants, researchers showed that slight differences in ZAP-70-mediated signaling strength affected arthritis susceptibility.132,133
For B cells, major defects in central tolerance leading to autoimmunity have been more difficult to prove and no AIRE-like equivalent has been discovered. Nevertheless, defective central tolerance of B cells in autoimmunity has been suggested by the finding of a higher frequency of naïve mature B cells with self-reactivity in patients with SLE and RA.134–136
Moving to the periphery, an example of defective peripheral tolerance promoting autoimmunity is FAS deficiency.137 FAS is a proapoptotic surface receptor that plays a critical role in maintaining immunologic homeostasis by eliminating undesired cells. Defects in FAS cause autoimmune lymphoproliferative syndrome (ALPS), also called Canale-Smith syndrome, and in mice, lymphoproliferative (lpr) disease, both of which exhibit massive enlargement of secondary lymphoid organs—caused mainly by the accumulation of a normally rare so-called “double negative” subset of T cells that lack both CD4 and CD8 co-receptors—and a variety of autoimmune manifestations. Lpr mice are defective in eliminating B cells that acquire self-reactivity in the periphery, a process that typically occurs in the GC during somatic hypermutation.129 Similar abnormalities have been described in humans and mice with defects in the ligand for FAS, FASLG.138
Peripheral tolerance is also breached by overexpression of the costimulatory molecule ICOS on T follicular helper (Tfh) cells in Sanroque mice. These mice have a point mutation in the Rc3h1 gene, a RING-type ubiquitin ligase, that impairs its ability to degrade ICOS mRNA.139 Increased expression of ICOS promotes the expansion of Tfh cells and germinal centers, production of IL-21, and autoimmunity.140
An example of a lymphocyte-extrinsic cause for loss of tolerance and autoimmunity is deficiency in the 3′ repair exonuclease 1 (TREX1, DNase III). Loss-of-function mutations in TREX1 have been implicated in the Aicardi-Goutières syndrome, a rare progressive encephalopathy associated with elevated IFN-α levels in cerebrospinal fluid, and chilblain lupus, a rare form of SLE characterized by painful, bluish-red inflammatory skin lesions typically affecting areas exposed to cold.141,142 Autoimmunity is thought to be caused by the intracellular accumulation of ssDNA derived from endogenous retroelements normally degraded by TREX1, resulting in the activation of intracellular DNA sensors and consequent over production of IFN-α.143,144 The importance of DNA disposal in lupus has been further illustrated by the association of DNase I deficiency with development of lupus in humans and mice.145,146
Defective regulation can also result in loss of tolerance and autoimmune disease as illustrated by the absence of Tregs.147,148 In humans, monogenic deficiency of the FOXP3 gene, which is required for Treg development, is associated with the IPEX (immune dysregulation, polyendocrinopathy, enteropathy X-linked) syndrome, a severe systemic autoimmune disease associated with diarrhea, eczematous dermatitis, and endocrinopathy, usually fatal within the first year. T1DM, autoimmune cytopenias, and nephritis are among other less common autoimmune manifestations of this syndrome. Similar findings are observed in scurfy mice that have a spontaneous function-impairing mutation of Foxp3.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


