56 Autoantibodies in Rheumatoid Arthritis
RFs have numerous causes in addition to RA and therefore have limited specificity for RA.
Autoantibodies targeting citrullinated proteins are hallmarks of the immune response in RA.
Rheumatoid Factor
The earliest assays (later called the Rose-Waaler agglutination test), which suggested the existence of autoantibodies in RA, were developed in the early 1940s. At this time serum from RA patients was shown to cause agglutination of sheep blood cells, which had been sensitized with subagglutinating doses of rabbit anti–sheep erythrocyte antibodies.1,2 These assays were later shown to detect immunoglobulin M (IgM) antibodies from RA patients that recognized the Fc portion of IgG3 (Table 56-1). Numerous modifications of the agglutination assay evolved, particularly the use of IgG-coated latex beads instead of sheep erythrocytes.4,5 Subsequent developments established RF assays in radioimmunoassay, enzyme-linked immunosorbent assay (ELISA), and nephelometry formats, with increased convenience but similar performance in terms of sensitivity and specificity. RFs may be positive in healthy controls (1% in younger individuals, up to 5% in individuals older than 70 years) and in patients with numerous other non-RA diseases (including other rheumatic diseases such as Sjögren’s syndrome and cryoglobulinemia), as well as chronic infections (see Table 56-1).6–8 The pretest probability of diagnosing RA therefore greatly influences the performance of the RF test, with sensitivities and specificities both in the 50% to 90% range, depending on the patient groups studied.9,10 The existence of IgG and IgA RFs and evidence of somatic hypermutation have suggested that some RFs in RA are T cell dependent.11,12 Although there have been studies suggesting that IgG and IgA RFs increase the specificity of RFs for RA,13 a recent meta-analysis showed that assays of the different RF isotypes performed similarly in terms of sensitivity and specificity and therefore do not add much over standard RF assays.14
Importantly, patients with RA also vary in terms of timing of RF appearance, with RFs preceding symptomatic disease in a significant subpopulation15–18 and following disease onset with variable kinetics in other patients. Indeed, the earlier onset of RF in patients with RA has been associated with more severe disease, highlighting a possible contribution of these antibodies to amplification.19 The observation that RF only appears after disease onset in some patients suggests that it may mark distinct, sequential events in pathogenesis that are close together in the subgroup in whom RF occurs early or are separated in time in patients in whom RF follows symptoms. The possible mechanisms whereby RF may play an amplifying role are numerous and include amplification of antigen capture, signaling, and effector functions, among others.20
Anticitrullinated Protein Autoantibodies
Antikeratin Antibodies and Antiperinuclear Factor in Rheumatoid Arthritis
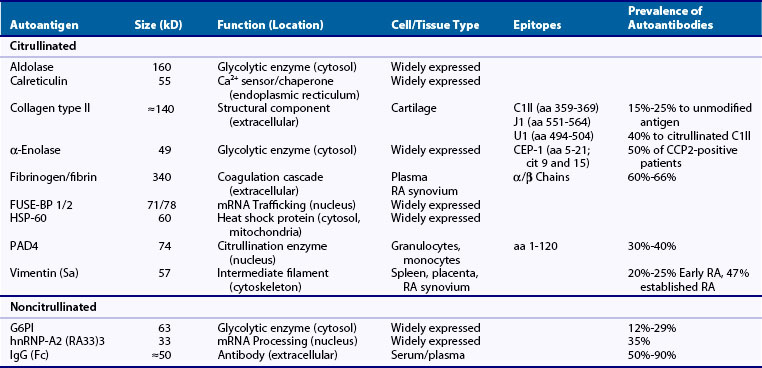
Although RFs have been clinically useful in diagnosis of RA, particularly in cases in whom the index of clinical suspicion is high, these autoantibodies have suffered from two shortcomings: (1) their lack of specificity for RA and (2) unclear kinetic and mechanistic connection to pathogenesis. Defining additional autoantibodies specific for RA has therefore been a major priority. A significant discovery in this regard was made initially by Nienhuis and Mandema in 196421 (Figure 56-1). They recognized an autoantibody that stained keratohyalin granules surrounding the nucleus in cells of human buccal mucosa, which they called antiperinuclear factor (APF). These APF antibodies were found in 49% to 91% of RA patients21–25; they have reported specificities of 73% to 99%.21,24–27 The assay was difficult to standardize because not every donor of buccal mucosal cells demonstrated the staining pattern.28–31 The assay therefore did not find its way into routine clinical practice, but the specificity for RA suggested that the assay might be reporting on a potentially important pathogenic pathway. A second research group subsequently demonstrated that RA sera recognized antigens in the stratified squamous layer of esophageal epithelium—a staining pattern that they termed antikeratin antibodies (AKAs).32 Subsequent iterations of this assay used other forms of stratified squamous epithelium including human skin.33 Although this AKA assay also had good specificity properties, it was of limited sensitivity34–37 and not particularly convenient or easy to standardize. Neither assay therefore was broadly used in the clinical diagnostic arena. Like the LE cell in SLE, however, the assays provided a framework for antigen discovery that would later lead to highly specific and convenient tools for RA diagnosis and classification.
Discovery of Autoantibodies That Recognize Peptidylcitrulline
The initial discovery of antibodies in RA patients, which recognized keratinized squamous epithelium, prompted studies to define the target of these antibodies. It was initially shown that APF only stained a fully differentiated squamous epithelial layer and that APF staining was identical to the pattern produced by a monoclonal antibody specific for human (pro)filaggrin in indirect immunofluoresence of permeabilized squamous epithelial cells23 (see Figure 56-1). Although the antigen could not be definitively identified using this co-localization alone, it suggested that a differentiation state might affect antigen recognition. Subsequently, RA sera were shown to recognize a 40-kD protein extracted from human epidermis; this was identified as a neutral/acidic isoform of filaggrin.38 IgG from RA sera affinity purified against the 40-kD filaggrin protein was reactive in both the APF and AKA tests, demonstrating that these autoantibodies were similar or identical.39 Filaggrin (filament-aggregating protein) is produced during the late stages of terminal differentiation of epithelial cells. It is synthesized as a heavily phosphorylated precursor protein (profilaggrin) that consists of 10 to 12 filaggrin repeats.40 Profilaggrin is deposited in granules, and it is released by proteolytic cleavage during differentiation of the cells, by proteases that remain to be fully defined. Coincident with cleavage, the protein is dephosphorylated and a significant proportion (≈20%) of the arginine residues are deiminated (i.e., converted to citrulline),41,42 a posttranslational modification mediated by the peptidylarginine deiminase (PAD) enzymes (see later).43 Because RA sera appeared to specifically target the neutral isoform of filaggrin (which is present and citrullinated in fully differentiated squamous epithelia), Schellekens and colleagues44 addressed whether RA sera positive in the AKA/APF assays specifically recognized citrullinated peptides. Thus regions within the deduced amino acid sequence of human profilaggrin with a high antigenicity index and the largest number of arginine residues were selected to generate synthetic peptides, where arginine residues were substituted with citrulline. Using these citrullinated peptides, Schellekens and colleagues demonstrated that AKA/APF antibodies are directed against citrullinated filaggrin. It is important to highlight that antibodies against citrullinated proteins in RA do not recognize free citrulline but do recognize citrulline residues (i.e., peptidylcitrullines) within the context of peptides or protein sequences.
These initial studies became the basis for the truly major discovery that protein sequences containing citrulline residues are one of the most prominent targets for autoantibodies in RA. However, because the epidermis is not a target of rheumatoid inflammation and there is no evidence that (pro) filaggrin is expressed in articular tissues, this molecule cannot be the autoantigen that drives the AKA/APF response in the joint. The facts that anticitrullinated filaggrin antibodies were enriched in synovial tissue compared with the serum or synovial fluid45 and that these antibodies are synthesized locally by plasmacytes within the rheumatoid pannus46 strongly suggest that anticitrullinated filaggrin cross-reacts with an antigen enriched in the RA joint/synovial tissue. Because citrullination is a frequent event in different tissues,47–51 great caution must be exercised when attempting to ascribe a primary role for a specific antigen in driving the antipeptidylcitrulline antibody response. The relevance of filaggrin in RA is therefore largely historical but unlikely of pathogenic relevance. Instead, its conformation and high content of citrulline residues make it an excellent surrogate with which to detect antibodies against citrullinated molecules. Many other citrullinated autoantigens found in the rheumatoid joint, which are currently being characterized, are much more likely to be of pathogenic relevance (see later).
Anti-CCP Antibodies: Clinical Relevance
In the initial characterization of AKA/APF antibodies, Schellekens and colleagues44 used a single C-terminal peptide derived from filaggrin (amino acids 306 to 324) to generate nine variants in which five arginine residues were changed to citrullines, either individually or in pairs, and these peptides were all assayed by ELISA. Interestingly, although the peptides were almost identical (the only difference being that the citrulline residues had different positions within the peptide), there were remarkable differences in the serum reactivity patterns toward each peptide (from 20% to 48% positivity), suggesting that although citrullination plays a critical role in antigen recognition by AKA/APF antibodies, the modification per se is not the only determinant that confers antibody binding. Instead, the data suggested that AKA/APF represent a pool of antibodies that recognize citrulline residues depending on the context of their surrounding amino acids. Indeed, when data from all peptides were pooled, the sensitivity increased to as much as 76%.44 Because peptides often adopt a β-turn conformation within the antibody-peptide complex52 and cysteine-bridged cyclic peptides have been shown to mimic the β-turn structure of the original antigenic determinant and can bind with enhanced affinity to antibodies,53 Schellekens and colleagues54 engineered a cyclic peptide (substituting the terminal serine residues with cysteines and cyclizing the peptide through the formation of a disulfide bond) to which ACPAs reacted with higher affinity (see Figure 56-1). Using this cyclic citrullinated peptide (later named CCP1) and its linear counterpart, it was demonstrated that the cyclic structure increased the sensitivity of the assay (68% vs. 49%, without affecting specificity),44,54 although it was still less sensitive than the assays using the combination of linear peptides (i.e., 76% vs. 68%).44 Nevertheless, CCP1 became the antigen for the first generation of ELISAs designed to detect antibodies against citrullinated autoantigens. To improve the CCP1 test, libraries of citrullinated peptides were used to construct the second-generation anti-CCP assay (CCP2), which was broadly adopted for clinical use.55,56 In 2005 a third generation of anti-CPP (CCP3) was made available for the laboratory diagnosis of RA. These assays have been reported to recognize additional citrullinated epitopes that are not identifiable with the second-generation CCP assays. In studies directly comparing second- and third-generation anti-CCP assays, the assays performed similarly,57,58 with a slightly increased sensitivity of CCP3 in some studies (e.g., sensitivities of 82.9% vs. 78.6% for CCP2, with specificities in the 93% to 94% range).58
The clinical importance of anti-CCP antibody tests in RA stems from several favorable features (Figure 56-2): (1) Anti-CCP antibodies have high specificity for the diagnosis of RA. In systematic reviews and meta-analyses, anti-CCP antibody positivity has been shown to be as sensitive as, but more specific than, RF in distinguishing RA from other forms of inflammatory arthritis.14 (2) The presence of anti-CCP autoantibodies is an important predictor for development of RA. More than 90% of the patients with undifferentiated arthritis who tested positive for anti-CCP develop RA within 3 years, in contrast to only 25% of the anti-CCP negative patients.59 (3) Anti-CCP positivity has been associated with a more severe and destructive disease course, although the independent role of the antibody in comparison with RF has not yet been confirmed. Some investigators have shown that both RF and anti-CCP antibodies are independent predictors of severity, whereas others have shown that anti-CCP antibodies rather than RF are the better predictor of radiographic progression,60–67 particularly in those patients who were seronegative for RF.67
The early and accurate diagnosis of RA, together with early, adequate use of disease-modifying antirheumatic drugs, decreases accrual of joint damage in RA. Because anti-CCP antibodies are a marker of disease severity and are detectable early in the disease course, they are powerful tools to classify patients with early inflammatory arthritis who will benefit from treatment.68–70
Kinetics of Appearance of Anti-CCP Antibodies in Rheumatoid Arthritis
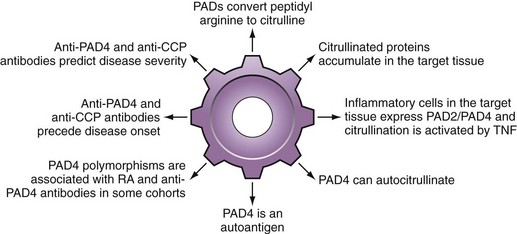
Recent studies have demonstrated that autoantibodies may precede onset of clinical disease in both tissue-specific and systemic autoimmune diseases.17,71–77 Defining events before clinical onset is challenging, and several different study designs have been employed to define the appearance of autoantibodies and their relationship with disease. The two general categories include (1) stored blood samples collected before disease onset (blood banks or military cohorts); (2) prospective studies examining emergence of autoantibodies and disease in high-risk individuals (often relatives of affected individuals).78 In RA, the former approach has clearly shown that autoantibodies precede RA in many individuals who subsequently develop seropositive RA, often by 2 to 6 years.76,77,79,80 In several studies, 20% to 60% of RA patients were RF positive before diagnosis and 30% to 60% of patients were positive for anti-CCP.76,77,79,80 With one exception,80 the prevalence of anti-CCP before diagnosis was almost twice as high as RF. Interestingly, although RF remained present consistently, anti-CCP could vary over time, even disappearing in some individuals. In one study, preclinical anti-CCP positivity was strongly associated with the development of erosive RA (odds ratio = 4.64; 95% confidence interval, 1.71 to 12.63; P < 0.01), whereas RF was not (P = 0.60).80 Together, these data highlight that the development of autoimmunity against citrullinated antigens is often asymptomatic and generally precedes the onset of clinical disease. This presymptomatic phase is potentially of great importance because it may identify individuals in whom important precursor conditions for the subsequent development of RA have been satisfied. The subsequent events that convert this RA precursor into a chronic, self-sustaining process are not yet known, but defining them is of great importance.
Anticitrullinated Protein Autoantibodies
Defining the primary antigen(s) that drive the generation of ACPAs has important implications in terms of RA etiology and pathogenesis. AKA/APF were the first ACPAs to be characterized, and they recognized citrullinated forms of filaggrin or its precursor profilaggrin.44,81
To date, several different approaches have been used to identify potential ACPA targets in RA. Although these approaches all use protein sequencing to identify autoantigens detected by ACPAs using immunoblotting assays, they vary in terms of the source of the antigens. Thus in addition to RA pannus, identification of citrullinated autoantigens has been extended to rheumatoid synovial fluid, as well as to cell lysates in which purified PADs have been used to generate in vitro citrullinated proteins. Using these approaches, several citrullinated autoantigen candidates have been identified. These include vimentin82,83; fibronectin; α-enolase, elongation factor-1α, and adenyl cyclase-associated protein-184; F-actin capping protein α-1-subunit, asporin, cathepsin D, beta-actin, histamine receptor, protein disulfide-isomerase, ER60 precursor, and mitochondrial aldehyde dehydrogenase85; collagen types I86 and II87; eukaryotic translation initiation factor-4G188; aldolase, phosphoglycerate kinase-1 (PGK1), calreticulin, HSP60, and the far upstream element binding proteins (FUSE-BP) 1 and 289; and PAD4.90 Interestingly, some of these molecules (e.g., vimentin, α-enolase, collagen type II, HSP60, aldolase, calreticulin, and PAD4) had been previously identified as autoantigens in RA, before recognizing that protein citrullination played a role in their recognition by autoantibodies84,91 (see Table 56-1). Although RA sera containing ACPAs may recognize both the native and the citrullinated forms of the antigen, the modified form is usually preferentially recognized. Among the citrullinated candidate antigens present in the joint, the best characterized clinically and pathogenically are fibrin(ogen), vimentin, collagen type II, and α-enolase. These antibodies are each dealt with briefly as follows.
Antibodies Recognizing Citrullinated Fibrinogen
The first local targets for ACPAs identified in RA synovial tissue were the citrullinated forms of the α and β chains of fibrin(ogen)92 (see Table 56-1). Using protein extracts from rheumatoid synovial membranes and affinity-purified anticitrullinated filaggrin antibodies, Masson-Bessière and colleagues92 identified for the first time that RA synovial tissue contains several citrullinated proteins and defined among them two proteins that were specifically targeted by anticitrullinated filaggrin antibodies. By amino-terminal sequencing the proteins were identified as the α- and β-chains of fibrin (although indeed, these sequences cannot allow distinguishing between fibrin and its precursor fibrinogen).92 Fibrin is the cleavage product of fibrinogen, a molecule that is processed by thrombin in the final steps of the coagulation cascade. Fibrinogen is a dimeric glycoprotein, with each monomer composed of three polypeptides: the Aα (610 residues), the Bβ (461 residues), and the γ (411 residues). Thrombin cleaves fibrinogen at Aα Arg-16 and at Bβ Arg-14, which result in the release of fibrinopeptides A and B, and exposure of the EA and EB polymerization sites. Cleavage of fibrinogen leads to formation of fibrin monomers that spontaneously polymerize to form a fibrin clot. Interestingly, fibrin deposition is usual during tissue inflammation and the presence of fibrin has long been known to be particularly abundant in rheumatoid synovial tissue,93 likely as a result of an altered balance between coagulation and fibrinolysis.93,94 Because circulating fibrinogen is not citrullinated, the presence of citrullinated fibrin in the rheumatoid synovium strongly suggested that after fibrin deposition, citrullination likely occurs in situ through the activity of locally expressed PAD. This proposal (i.e., that the joint is the site for autoantigen citrullination in RA) has been further supported and extended to many other citrullinated autoantigens in RA.95 Citrullinated fibrinogen antibodies are among the most frequent ACPAs found in patients with RA (≈60% to 66%).96–98 In total, 54 of the 81 arginines (66%) in human fibrinogen have been found to be susceptible to citrullination.99–101 Using linear citrullinated peptides, major citrullinated epitope regions have been identified in this molecule, with some overlap among studies, but also with clear differences.101,102 Although some of these peptides have been used to detect ACPAs against citrullinated fibrinogen, whole citrullinated fibrinogen is the more widely used antigen in these assays.96–98
Antibodies Recognizing Citrullinated Vimentin
The Sa autoantigen (named after a patient, Mrs Sa …) was described between 1992 and 1994 as an approximately 50-kD band detected by immunoblotting using RA sera against normal human spleen, placenta, and rheumatoid synovial extracts.103,104 Subsequent studies confirmed that anti-Sa antibodies are highly specific for RA (≈95%), with a sensitivity that varies with the stage of the disease tested, ranging from 20% to 25% in early RA cohorts and 47% in patients with more established disease.104–106 However, although anti-Sa autoantibodies were shown to be present early in the disease and to be markers of an aggressive and destructive form of RA,105 their diagnostic value was limited because of the challenge in standardizing and hence marketing the assay. Further attempts to purify the Sa antigen from placenta identified vimentin as the elusive candidate,83,107 a molecule that was known to be citrullinated in activated macrophages108 (see Table 56-1). Vimentin is an intermediate filament protein important in the dynamic organization of the cytoskeleton, with a vital function in organelle transport, cell migration, and proliferation.109,110 Vimentin filaments are also involved in the regulation of mechanical stress between chondrocytes and the surrounding matrix tissues. Its assembly and disassembly are regulated by phosphorylation and potentially by deimination.111 During macrophage apoptosis, vimentin is citrullinated and this process has been suggested to be involved in filament disassembly.108 Whether citrullination of vimentin is involved in other physiologic processes is unknown. The frequency of anticitrullinated vimentin antibodies in RA varies widely depending on the study and the antigen used for detection. Initially, partially purified preparation of placental Sa antigen was used to detect anti-Sa antibodies by Western blot. For this assay, the sensitivity varies from 22% in patients with early arthritis to 47% in patients with established disease.104–106 More recently, Bang and colleagues112 identified various citrullinated and mutated isoforms of vimentin in synovial fluid of RA patients and suggested that mutations in vimentin may result from oxidative stress–induced DNA damage in RA synovium. Protein sequence data demonstrated that the vimentin isoforms contained certain amino acid mutations and modifications, in particular mutations of glycine residues into arginine residues at positions 16, 59, 145, and 147, allowing novel citrullination in the mutated residues 145 and 147. Using this molecule (named mutated citrullinated vimentin or MCV) in ELISA assays, the sensitivity of ACPAs (presumably against citrullinated vimentin) increased to 54% in patients with early synovitis who developed RA113 and as high as 92% in patients with early RA (<2 years’ duration) versus 81% in patients with established RA (>2 years’ duration).114 The overall sensitivity and specificity profile of this assay has been noted to be the best among available ACPAs (i.e., around 84% and 87%).115
Antibodies Recognizing Citrullinated Enolase
Antibodies against the immunodominant epitope of α-enolase, citrullinated α-enolase peptide 1 (CEP-1),116 occur in approximately half of anti-CCP2-positive patients117 (see Table 56-1). Enolase, also known as phosphopyruvate dehydratase, is a metalloenzyme responsible for the catalysis of the conversion of 2-phosphoglycerate to phosphoenolpyruvate, the penultimate step of glycolysis. It is also involved in various processes including hypoxia tolerance, growth control, and fibrinolysis.118 There are three subunits of enolase, α, β, and γ, each encoded by a separate gene that can combine to form five different isoenzymes: αα, αβ, αγ, ββ, and γγ. Three of these isoenzymes (all homodimers) are more commonly found in adult human cells than the others: αα or non-neuronal enolase (also known as enolase 1) is found in a variety of tissues including liver, brain, kidney, spleen, and adipose; ββ or muscle-specific enolase (enolase 3); and γγ or neuron-specific enolase (enolase 2). Citrullinated α-enolase is a target for ACPAs.84 However, whether this modification plays a role in the physiologic function of the enzyme or whether its citrullination only occurs under pathologic conditions (e.g., during cell death, either intracelluarly or extracellularly) remains unclear. Using cyclic citrullinated α-enolase peptides covering 15 out of 17 arginine residues present in human α-enolase, CEP-1 was identified as the immunodominant B-cell epitope.116 This region comprised amino acids 5 to 21 of α-enolase, with arginine-9 and arginine-15 replaced by citrulline. Although antibodies against CEP-1 have not shown a better diagnostic or prognostic value than anti-CCP2,117 α-enolase has gained special attention as a potential target for disease initiation (see later).
Antibodies Recognizing Citrullinated Collagen Type II
Type II collagen is the predominant collagenous component of cartilage and is an autoantigen implicated in disease initiation in RA (see Table 56-1). Immunization with collagen type II is associated with development of autoimmune arthritis in several species, and the epitope-specific recognition of type II collagen by RA antibodies is shared by antibodies that are arthritogenic in collagen-induced arthritis (CIA) in the mouse.119 The antibody response to collagen type II is predominantly directed toward conformational triple-helical structures, and the identification of relevant B-cell epitopes has required the construction of recombinant triple-helical proteins and synthetic triple-helical peptides.120 The major B-cell epitopes on triple-helical collagen type II identified including C1 III (amino acids 359 to 369), J1 (amino acids 551 to 564), and U1 (amino acids 494 to 504) seem to share a common motif, comprising an arginine–glycine-hydrophobic amino acid motif. The observation that arginines occur in most of the collagen type II epitopes has raised the possibility that these may be citrullinated by PADs and that autoantibodies might bind specifically to the citrullinated forms. Indeed, using triple-helical molecules with the C1 epitope citrullinated, the diagnostic sensitivity for RA was 40%,87,98 compared with only 15% to 25% using the unmodified antigen.87,121–124
Citrullinated Antigen Generation in Rheumatoid Arthritis
Peptidylarginine Deiminase Enzymes
The PADs, which are deiminating enzymes that hydrolyze guanidinium side chains in peptidylarginine to yield peptidylcitrulline and ammonia, belong to a larger group of guanidino-modifying enzymes called the amidinotransferase (AT) superfamily.125,126 Additional members of this superfamily include (1) the arginine deiminases (ADIs), enzymes found in both prokaryotes and the primitive eukaryote Giardia intestinalis, that act on nonpeptidyl arginine and that are involved in energy production; (2) the dimethylarginine dimethylaminohydrolases (DDAHs), enzymes found in both bacteria and mammals that convert nonpeptidyl methylarginines into citrulline; (3) the ATs, enzymes involved in both creatine and streptomycin biosynthesis; and (4) the dihydrolases, bacterial enzymes involved in arginine catabolism that catalyze two successive hydrolytic steps.126 Whereas most superfamily members act on nonpeptidyl amino acids, PADs are highly specific for peptidylarginine residues and require at least one additional residue N-terminal to the site of modification.127 Although the chemical reaction catalyzed by PAD enzymes is officially called peptidylarginine deimination, the term citrullination is more generally used. PAD and PAD-like enzymes are present in numerous species, from bacteria to humans. Interestingly, the bacterial PADs are composed of only the approximately 40-kD catalytic domain, whereas mammals have larger multidomain enzymes (≈75 kD), whose activity is regulated by calcium. The extra N-terminal domains in the mammalian enzymes include two immunoglobulin-like domains that are proposed to mediate protein–protein interactions and/or substrate targeting.128 To date, five human PAD homologs have been identified and the PADI genes are located at a single cluster on chromosome 1p36.1.43,129 For historical reasons, these isozymes are designated PAD1-4 and PAD6. Human PAD4 was initially named PAD5 but was later renamed PAD4 to reflect the fact that it is a true ortholog of this isoform.130 PAD4 is the only PAD that resides in the nucleus, a function of its nuclear localization sequence.131
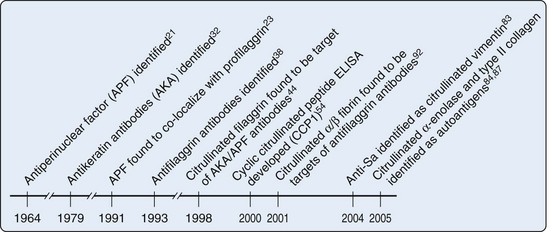
The PADs share 50% to 55% sequence identity with each other,128 with different PADs preferentially expressed in specific tissues. PAD1 is primarily expressed in uterus and skin. PAD2 is more widely expressed in muscle, skin, brain, spleen, rheumatoid synovium, and secretory glands. PAD3 is also expressed in skin, PAD4 is expressed in hematopoietic cells, and PAD6 is found in germ cells and peripheral blood leukocytes.43,82,129,130,132–134 Because of their prominent expression in rheumatoid synovial tissue and fluid,95,135,136 PAD2 and PAD4 have gained prominence as potential candidates that drive citrullination of self-antigens in RA.
Peptidylarginine Deiminase Structure, Activity, and Regulation
The three-dimensional structure of PAD4 has been solved128,137 and is likely representative of the broader family. PAD4 is a dimer formed by head-to-tail contact between the N-terminal domain of one molecule and the C-terminal domain of the second. Each monomer adopts an elongated fold in which the N-terminal domain forms two immunoglobulin-like subdomains (named subdomain 1 and 2), and the C-terminal domain forms an α/β propeller structure. Five Ca2+-binding sites were identified in the structure, and Ca2+ binding induces conformational changes required to generate the active site cleft. Thus PADs rely strongly on the presence of Ca2+ for activity and require millimolar amounts of calcium to be activated in vitro. Because calcium concentrations in cells do not rise above the low micromolar levels, the mechanisms that control PAD activation in vivo remain unclear. One possible explanation for the requirement of “extracellular” concentrations of calcium to achieve PAD activation is that protein citrullination occurs under extreme conditions where intracellular concentrations of Ca2+ approach extracellular concentrations (e.g., during necrosis or apoptosis).108 Whether other mechanisms regulate PAD enzyme activation, potentially by modifying the calcium requirement (e.g., allosteric effects of post-translational modifications or partner-binding), is not known. It is also important to note that although PAD activation provides one level of regulation, the activity of the PADs can also be negatively regulated through autocitrullination. This is not dissimilar to other enzymes, where automodification (e.g., autophosphorylation) modifies enzymatic function. Autocitrullination has been described for PADs 1, 2, 3, and 4.90,138 In the case of PAD4, direct citrullination of arginines surrounding the active site cleft appears to have a major impact on activity.90
Structural and Functional Implications of Protein Citrullination
Arginine residues often play a central role in the maintenance of tertiary structure in proteins because the positively charged guanidino group is a versatile interacting partner, forming multiple intramolecular hydrogen bonds to backbone carbonyl oxygens, and also intermolecularly between different proteins.139 Thus during the process of citrullination, the conversion of arginine into citrulline reduces the net charge of the protein by the loss of a positive charge per citrulline residue. In addition, by changing the guanidino group in arginine to an ureido group in citrulline, arginine deamination may modify intramolecular and intermolecular interactions, producing changes in protein structure.140
These structural changes likely also have important functional implications. Indeed, deimination has been implicated in several physiologic processes. In the skin (which expresses PADs 1, 2, and 348,133,141), deimination of filaggrin (a component of the cornified cell envelope) is critical for its degradation into free amino acids, which act as a natural moisturizing factor in the stratum corneum.48,141 Interestingly, PAD2-deficient mice have normal development and show no abnormalities in the skin,142 suggesting functional redundancy in mouse development. Whether PAD2 functions in the skin during injury or environmental perturbations is currently unknown. In the immune system, PAD2 (and/or potentially other PADs) citrullinates chemokines, a process that decreases chemokine activity.143–146 Thus PAD2 may play important roles in controlling effector functions related to environmental triggers.
Human PAD4 (first called PAD5) was first found in human myeloid leukemia HL-60 cells induced to differentiate into granulocytes by retinoic acid130 and later described in peripheral blood granulocytes.132 The expression of PAD4 during granulocyte differentiation initially suggested that this enzyme may have a role during granulopoiesis. Histones H2A, H3, and H4, as well as nucleophosmin/B23, were the first PAD4 substrates to be identified.147 PAD4 appears to function as a transcriptional co-regulator, mediated by its ability to catalyze the deimination of specific residues present in the N-terminal tails of histones.148–150 Interestingly, mice deficient in PAD4 also have normal development,151 suggesting that this enzyme has no essential roles in steady-state cellular functions and/or development. Recent studies have demonstrated that PAD4-mediated citrullination of histones is required for NET (neutrophil extracellular traps) formation and bacterial clearance,150,151 suggesting a role for this enzyme in immune and inflammatory effector functions.
PAD6, the most recently defined member of the PAD family, was initially cloned from mouse oocytes and named egg PAD (ePAD).152 PAD6 is essential for the formation of cytoplasmic lattices, and female mice deficient in PAD6 are infertile but otherwise normal.134
Peptidylarginine Deiminases in Rheumatoid Arthritis
Multiple PADs are likely responsible for autoantigen citrullination in RA, particularly PADs 2 and 495,135,136 (see Figure 56-2). Although PAD4 has some unique characteristics that have focused significant attention on this protein, it is important to be aware that there is no evidence to suggest that pathologic citrullination in RA is exclusively or preferentially mediated by this isoform.
It is of interest that several polymorphisms in PAD4 have been genetically associated with RA development, particularly in Asian populations153–156 (see Figure 56-2). In this regard, two common haplotypes of the PADI4 gene were initially identified153 and designated ”susceptible” or “nonsusceptible” on the basis of their relative frequency in patients with RA versus controls. In the initial population, the odds ratio for the susceptible haplotype was almost 2; this effect was not observed in most Caucasian populations. Changes in mRNA stability of the susceptible PADI4 reported in the initial paper were not associated with changes in enzyme level. Furthermore, although the susceptible haplotype generates a PAD4 molecule containing three amino acid substitutions in the N-terminal region (i.e., Gly55-Ser, Val82-Ala, Gly112-Ala), current evidence suggests that these changes have no effect on the function of the protein.90,157 Indeed, they appear to affect conformation only within the N-terminal domain, but not the active site located in the C-terminal domain.157 The fact that PAD4 acts as a head-to-tail dimer may nevertheless allow conformational differences in the N-terminus of one molecule to influence the C-terminal domain of another and influence catalysis or even inhibition by autocitrullination. It has been proposed that PAD4 genotype may exert its effects on RA disease susceptibility as a consequence of its immunogenicity, rather than its enzyme activity.90
Consistent with this hypothesis is the observation that, in addition to its ability to citrullinate RA autoantigens, PAD4 itself is an RA autoantigen, even in the noncitrullinated form158–160 (see Table 56-1 and Figure 56-2). The sensitivity of PAD4 antibodies for RA is in the 30% to 40% range, with specificity of greater than 95%. PAD4 autoantibodies are associated with more severe, erosive RA that persists despite treatment with tumor necrosis factor (TNF) inhibitors160,161 and, like anti-CCP, are frequently present early before onset of any symptoms.74 Interestingly, the susceptible PAD4 haplotype was strikingly associated with PAD4 autoantibodies, even in Caucasian populations in whom the RA disease association was not evident.159
Genetic Associations with Anti-Ccp/Acpas
Although the association of specific HLA-DR alleles and RA has been known for decades, the relationship between genetics and the development of RA autoantibodies is just beginning to be elucidated.162 A subset of HLA-DR alleles termed the “shared epitope” (SE) alleles includes HLA-DR*0101, *0102, *0401, *0404, *0405, *0408, *0410, *1001, and *1402 and is united by a conserved sequence of amino acids (QRRAA, QKRAA, or RRRAA) on the α-helix of the DR-β chain peptide binding groove.163 SE alleles are strongly associated with the development of anti-CCP/ACPA but are not independently associated with RF59 (Figure 56-3). Furthermore, there appears to be a gene dosage effect on the relative risk of anti-CCP development with an odds ratio of 3.3 to 4.7 for patients with one SE allele and an odds ratio of 11.8 to 13.3 for patients with two SE alleles.59,164,165 Analysis of individual SE alleles has revealed that anti-CCP/ACPAs are predominantly associated with HLA-DR4 rather than HLA-DR1 SE alleles.98 Interestingly, smoking has been shown to skew the genetic association of SE alleles with anti-CCP development, with HLA-DR1 and HLA-DR10 SE alleles being more important in this patient group (see “Autoantibodies in Rheumatoid Arthritis: Insights into Disease Mechanism” later).166
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


