82 Antiphospholipid Syndrome
The clinical manifestations range from asymptomatic to catastrophic antiphospholipid syndrome (APS).
Diagnosis of the antiphospholipid syndrome (APS) requires that a patient have both a clinical event (thrombosis or pregnancy morbidity) and the persistent presence of antiphospholipid antibody (aPL), documented by a solid phase serum assay (anticardiolipin or anti–β2-glycoprotein I [anti-β2GPI] immunoglobulin [Ig]G or IgM), a coagulation assay (inhibitor of phospholipid-dependent clotting—the lupus anticoagulant test), or both. Preliminary (Sapporo) classification criteria for APS,1 revised in 2004,2 are listed in Table 82-1.
Table 82-1 Revised Sapporo Classification Criteria for Antiphospholipid Syndrome
Investigators are strongly advised to classify APS patients participating in studies into one of the following categories: I, more than one laboratory criteria present (any combination); IIa, only lupus anticoagulant present; IIb, only anticardiolipin antibody present; IIc, only anti–β2-glycoprotein I antibody present.
APS, antiphospholipid syndrome; ELISA, enzyme-linked immunosorbent assay.
* Coexisting inherited or acquired factors for thrombosis are not reasons to exclude patients from APS trials. However, two subgroups of APS patients should be recognized by the presence and absence of additional risk factors for thrombosis. Indicative (but not exhaustive) cases include age (>55 yr in men and >65 yr in women), the presence of any of the established risk factors for cardiovascular disease (hypertension, diabetes mellitus, elevated low-density lipoprotein or low high-density lipoprotein cholesterol, cigarette smoking, family history of premature cardiovascular disease, body mass index >30 kg/m2, microalbuminuria, estimated glomerular filtration rate <60 mL/min), inherited thrombophilias, oral contraceptive use, nephritic syndrome, malignancy, immobilization, and surgery. Thus, patients who fulfill these criteria should be stratified according to contributing causes of thrombosis.
† A thrombotic episode in the past can be considered a clinical criterion, provided that thrombosis is proved by appropriate diagnostic means, and that no alternative diagnosis or cause of thrombosis is found.
‡ Superficial venous thrombosis is not included in the clinical criteria.
§ Generally accepted features of placental insufficiency include an abnormal or nonreassuring fetal surveillance test (e.g., nonreactive nonstress test) suggestive of fetal hypoxemia, an abnormal Doppler flow velocimetry waveform analysis suggestive of fetal hypoxemia (e.g., absent end-diastolic flow in the umbilical artery), oligohydramnios (e.g., amniotic fluid index ≤5 cm), or a postnatal birth weight less than the 10th percentile for gestational age.
From Miyakis S, Lockshin MD, Atsumi T, et al: International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome, J Thromb Haemost 4:295–306, 2006.
Certain factors are not included as criteria but may be helpful in the diagnosis of individual patients. These include IgA anticardiolipin or anti-β2GPI, valvular heart disease, thrombocytopenia, early preeclampsia, and livedo reticularis (Table 82-2). These factors are rare, nonstandardized, or nonspecific phenomena that are too unreliable for use in clinical studies, but they occur in a sufficient number of patients to support a suspected diagnosis.
Table 82-2 Other Features Suggesting the Presence of Antiphospholipid Antibodies
| Clinical |
| Laboratory |
APS can occur as an isolated diagnosis, or it can be associated with systemic lupus erythematosus (SLE) or another rheumatic disease. Transient aPL, but probably not the syndrome, can be induced by drugs and infection.3
Epidemiology
Low-titer, usually transient, anticardiolipin occurs in up to 10% of normal blood donors,4,5 and moderate- to high-titer anticardiolipin or a positive lupus anticoagulant test occurs in less than 1%. The prevalence of positive aPL tests increases with age. Ten percent to 40% of SLE patients5 and approximately 20% of rheumatoid arthritis patients6 have positive aPL tests.
Based on a limited number of uncontrolled and non–risk-stratified studies, asymptomatic (no history of vascular or pregnancy events) aPL-positive patients have a 0% to 4% annual risk of thrombosis; patients with other autoimmune diseases such as SLE are at the higher end of the range.7,8 The aPL profile (low vs. high risk for thrombosis) and patients’ clinical characteristics (presence or absence of other acquired or genetic thrombosis risk factors) influence the individual risk of thrombosis.9 Ten percent of first-stroke victims have aPLs,10 especially those who are young (up to 29%),5,11 as do up to 20% of women who have suffered three or more consecutive fetal losses.12 Fourteen percent of patients with recurrent venous thromboembolic disease have aPLs.13
Cause
The main antigen to which aPLs bind is not a phospholipid but rather a phospholipid-binding plasma protein, namely, β2GPI (apolipoprotein H). β2GPI is normally present in serum at a concentration of 200 mg/mL, is a member of the complement control protein family, and has five repeating domains and several alleles. An octapeptide in the fifth domain and critical cysteine bonds are necessary for both phospholipid binding and antigenicity14; a first-domain site activates platelets.15,16 In vivo, β2GPI binds to phosphatidylserine on activated or apoptotic cell membranes, including those of trophoblasts, platelets, and endothelial cells. Under physiologic conditions, β2GPI may function in the elimination of apoptotic cells17 and as a natural anticoagulant.18
Other, less relevant antigens targeted by aPLs are prothrombin, annexin V, protein C, protein S, high- and low-molecular-weight kininogens, tissue plasminogen activator, factor VII, factor XI, factor XII, complement component C4, and complement factor H.19
In experimental animal models, passive or active immunization with viral peptides,20 bacterial peptides,21 and heterologous β2GPI22 induces polyclonal aPLs and clinical events associated with APS. These data suggest that pathologic autoimmune aPL is induced in susceptible humans by infection via molecular mimicry.
However, infection-induced aPLs (syphilitic and nonsyphilitic Treponema, Borrelia burgdorferi, human immunodeficiency virus, Leptospira, or parasites) are usually β2GPI independent and bind phospholipids directly.23 Drugs (chlorpromazine, procainamide, quinidine, and phenytoin) and malignancies (lymphoproliferative disorders) can also induce β2GPI-independent aPLs. Conversely, autoimmune aPLs bind β2GPI or other phospholipid-binding plasma proteins, which in turn bind negatively charged phospholipids such as cardiolipin (β2GPI-dependent aPLs).
Pathogenesis
Antiphospholipid antibody is most likely related to thrombosis through multiple mechanisms; a proposed pathogenesis is illustrated in Figure 82-1. The process begins with activation or apoptosis of platelets, endothelial cells, or trophoblasts, during which phosphatidylserine (a negatively charged phospholipid) migrates from the inner to the normally electrically neutral outer cell membrane. Circulating β2GPI binds to phosphatidylserine, and then aPL binds to a β2GPI dimer.24
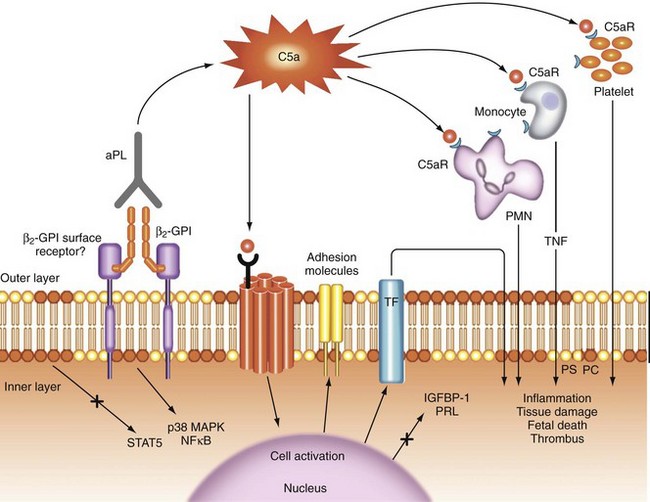
Antiphospholipid antibody–β2GPI dimer binding activates the complement cascade extracellularly; initiates an intracellular signaling cascade, probably through the C5a and β2GPI surface receptors; and recruits and activates inflammatory effector cells, including monocytes, neutrophils, and platelets, leading to the release of proinflammatory products (e.g., tumor necrosis factor [TNF], oxidants, proteases) and the induction of a prothrombotic phenotype.25–27 The putative receptor of β2GPI binding protein that transduces signals from the cell membrane to the nucleus is not yet identified and may vary among cells. The following candidates have been suggested: apoER2 (a member of the low-density lipoprotein receptor superfamily), annexin A2, and a Toll-like receptor.28–30 Both nuclear factor κB and p38 mitogen-activated protein kinase may play a role in the intracellular signaling cascade.31,32
In addition, through downregulation of the signal transducer and activator of transcription 5 (STAT5), aPLs inhibit the production of placental prolactin and insulin growth factor–binding protein-1,33 and they adversely affect the formation of a trophoblast syncytium, placental apoptosis, and trophoblast invasion—all processes that are required for the normal establishment of placental function.
Other possible contributory mechanisms of aPL-mediated thrombosis include inhibition of coagulation cascade reactions catalyzed by phospholipids (e.g., activation of circulating procoagulant proteins, inhibition of protein C and S activation), induction of tissue factor (a physiologic initiator of coagulation) expression on monocytes, reduction of fibrinolysis, and interaction with the annexin V anticoagulant shield in the placenta.29
In experimental animal models, aPLs cause fetal resorption and increase the size and duration of trauma-induced venous and arterial thrombi.34,35 Inhibiting complement activation prevents experimental aPL-induced fetal death and angiogenic dysregulation–associated abnormal placental development and preeclampsia; C5 knockout mice carry pregnancies normally despite aPL,36 implying that a complement-mediated effector mechanism is an absolute requirement for fetal death to occur. Complement activation is also required for experimental thrombosis.37,38 In addition, aPL-induced reduction of heparin-binding epidermal growth factor–like growth factor leads to defective placentation.39
Because high-level aPLs may persist for years in asymptomatic persons, it is likely that vascular injury, endothelial cell activation, or both immediately precede the occurrence of thrombosis in those bearing the antibody (second-hit hypothesis). Of note, at least 50% of APS patients with vascular factors possess other acquired thrombosis risk factors at the time of their events.40,41
Persons congenitally lacking β2GPI42 and β2GPI knockout mice appear normal.43 β2GPI polymorphisms influence the generation of aPLs in individuals, but they have only a weak relationship to the occurrence of APS.44 A cluster of 50 upregulated genes may have an effect on the occurrence of thrombosis in aPL-positive individuals.45
Clinical Features
Vascular Occlusion
APS affects all organ systems. Its principal manifestations are venous or arterial thromboses and pregnancy loss (see Table 82-1). Except for their severity, the youth of affected patients, and the unusual anatomic locations (Budd-Chiari syndrome; sagittal sinus and upper extremity thromboses), venous thromboses in APS do not differ clinically from thromboses attributable to other causes. Similarly, arterial thromboses differ from non–aPL-associated thromboses only by their recurrent nature, unusual locations, and occurrence in young patients. Deep vein thrombosis and stroke are the most common clinical manifestations of APS. Renal thrombotic microangiopathy, glomerular capillary endothelial cell injury, and thrombosis of renal vessels cause proteinuria without cells in the urine or hypocomplementemia and may lead to severe hypertension, renal failure, or both.46
Pregnancy Morbidity
Pregnancy losses in patients with aPLs typically occur after 10 weeks’ gestation (fetal loss), although earlier losses also occur. These pre-embryonic or embryonic pregnancy losses (<10 weeks’ gestation) are commonly due to chromosomal and other genetic defects. Pregnancy in those with APS is often normal until the second trimester, when fetal growth slows and amniotic fluid volume decreases. APS patients may develop severe, early preeclampsia or HELLP (hemolysis, elevated liver enzymes, low platelets) syndrome. Placental infarction is a cause of fetal growth restriction or death; nonthrombotic mechanisms of placental dysfunction also may occur.47 Prior late pregnancy losses predict future losses, independent of the aPL profile.
Miscellaneous and Noncriteria Manifestations
Many patients have livedo reticularis or thrombocytopenia (Figure 82-2), although these conditions are not specific for APS. Cardiac valve disease (vegetations, thickening, or both), a late manifestation, may necessitate valve replacement. Its pathogenesis in APS is unknown. Recent studies suggest that APS does not add to the risk of atherosclerosis imparted by SLE.48 Pulmonary hypertension may develop owing to recurrent pulmonary embolism or small vessel thrombosis; rarely, aPL-positive patients may present with diffuse pulmonary hemorrhage. Some patients develop nonfocal neurologic symptoms such as lack of concentration, forgetfulness, and dizzy spells. Multiple small, hyperintense lesions seen on magnetic resonance imaging (MRI), primarily in the periventricular white matter, do not correlate well with clinical symptoms. Rarely, high-affinity antiprothrombin antibodies may cause hemorrhage by depleting prothrombin (lupus anticoagulant hypoprothrombinemia syndrome).49
Catastrophic Antiphospholipid Syndrome
Catastrophic APS is a rare, abrupt, life-threatening complication. It consists of multiple thromboses of medium and small arteries occurring (despite apparently adequate anticoagulation) over a period of days and causing stroke; cardiac, hepatic, adrenal, renal, and intestinal infarction; and peripheral gangrene.4,50 In a review of 220 patients with catastrophic APS, the main clinical manifestations included renal involvement in 154 patients (70%), pulmonary in 146 (66%), cerebral in 133 (60%), cardiac in 115 (52%), and cutaneous in 104 (47%).51 Acute adrenal failure may be the initial clinical event. Proposed formal criteria for this syndrome are shown in Table 82-3.52 Patients often have moderate thrombocytopenia; erythrocytes are less fragmented than in the hemolytic uremic syndrome or thrombotic thrombocytopenic purpura, and fibrin split products are not strikingly elevated. Renal failure and pulmonary hemorrhage may occur. Tissue biopsies show noninflammatory vascular occlusion.
Table 82-3 Preliminary Criteria for the Classification of Catastrophic Antiphospholipid Syndrome (APS)








