Chapter 77 Tumors and Tumorous Conditions of the Hand
Classification
Tumors involving the hand are classified in a manner similar to that for tumors involving the rest of the body. Benign tumors are classified as latent, active, or aggressive, according to their local biological activity (Table 77-1). Benign latent tumors are those in which tumor growth has occurred during childhood or adolescence and has subsequently entered an inactive or healing phase. Solitary and unicameral bone cysts are examples of benign latent tumors. A benign active tumor continues to enlarge and, although it is well encapsulated, may have an irregular or lumpy border. Most benign tumors of the hand fall into this category. A benign aggressive tumor, although nonmetastasizing and innocent appearing on histological sections, is locally destructive and is surrounded by a thin, tenuous capsule that may not contain all the tumor cells. A giant cell tumor of bone often behaves in this aggressive manner. A wide margin often is necessary for complete eradication of benign aggressive tumors.
TABLE 77-1 Classification of Benign Tumors
| STAGE | TYPE |
|---|---|
| 1 | Latent |
| 2 | Active |
| 3 | Aggressive |
Modified from Enneking WE: Musculoskeletal tumor surgery, New York, 1983, Churchill Livingstone.
Malignant tumors are classified as low grade (I), high grade (II), or associated with metastasis (III) (Table 77-2). Most malignant tumors of the hand are low-grade tumors and are classified further, according to the degree of local extension, as either intracompartmental (A) or extracompartmental (B). In the hand, each ray forms a distinct compartment. The individual phalanges are not considered separate compartments, but rather they, along with their corresponding intrinsic muscles, are included in the ray compartment. The ray compartment includes the flexor tendon and sheath of each finger as far proximally as the midpalmar space and the extensor tendon as far as the metacarpophalangeal joint. Each metacarpal is a separate compartment. If a tumor involves the palmar space or the loose areolar tissue on the dorsum of the hand, it is considered extracompartmental because proximal spread is unobstructed. Tumors arising in the digits remain confined to that compartment for long periods and then extend into the palm.
TABLE 77-2 Classification of Malignant Tumors
| STAGE | TYPE |
|---|---|
| IA | Low grade, intracompartmental |
| IB | Low grade, extracompartmental |
| IIA | High grade, intracompartmental |
| IIB | High grade, extracompartmental |
| III | Either grade with regional or distant metastasis |
Modified from Enneking WF, Spanier SS, Goodman MA: The surgical staging of musculoskeletal sarcoma, Clin Orthop Relat Res 153:106, 1980.
Treatment
Generally, hand tumors are treated surgically. Biopsy rarely is needed in most tumors because complete excision usually is indicated, and the entire tumor is available for microscopic study. Incisional biopsy is advised if a malignant tumor is suspected, or if the morbidity of surgical excision outweighs the morbidity caused by the tumor itself, as may be true in some benign neural tumors. Incisions should be made directly over the mass to be harvested for biopsy and should be oriented so as not to jeopardize hand function or interfere with complete removal of the tumor. The way in which a tumor is removed depends on its location, aggressiveness, potential for metastasizing, and, at times, sensitivity to adjuvant chemotherapy and radiation therapy. The various surgical margins are summarized in Table 77-3.
TABLE 77-3 Classification of Surgical Margins
| TYPE | PLANE OF DISSECTION |
|---|---|
| Intracapsular | Piecemeal, debulking, or curettage |
| Marginal | Shell out (en bloc) through pseudocapsule or reactive zone |
| Wide | Intracompartmental (en bloc) with cuff of normal tissue |
| Radical | Extracompartmental (en bloc) with entire compartment |
Modified from Enneking WE: Musculoskeletal tumor surgery, New York, 1983, Churchill Livingstone.
Benign Tumors (Table 77-4)
Lipoma
Although lipomas are more common elsewhere in the body, they also are found in the hand and are common solid cellular hand tumors (Fig. 77-1). These lightly encapsulated tumors are composed of mature fatty tissue in which the central lipid droplet and peripherally located nucleus form the characteristic signet-ring cell. They arise from mesenchymal primordial fatty tissue cells. These tumors can be superficial, arising from the subcutaneous tissues and having the characteristic signs of a soft, fluctuant, bulging mass, or they can occur deep in the palm, arising within the Guyon canal, the carpal tunnel, or the deep palmar space. Usually, a painless mass is present that impairs grasp. Lateral deviation of the fingers also may be present when the tumor is located around the metacarpophalangeal joints.
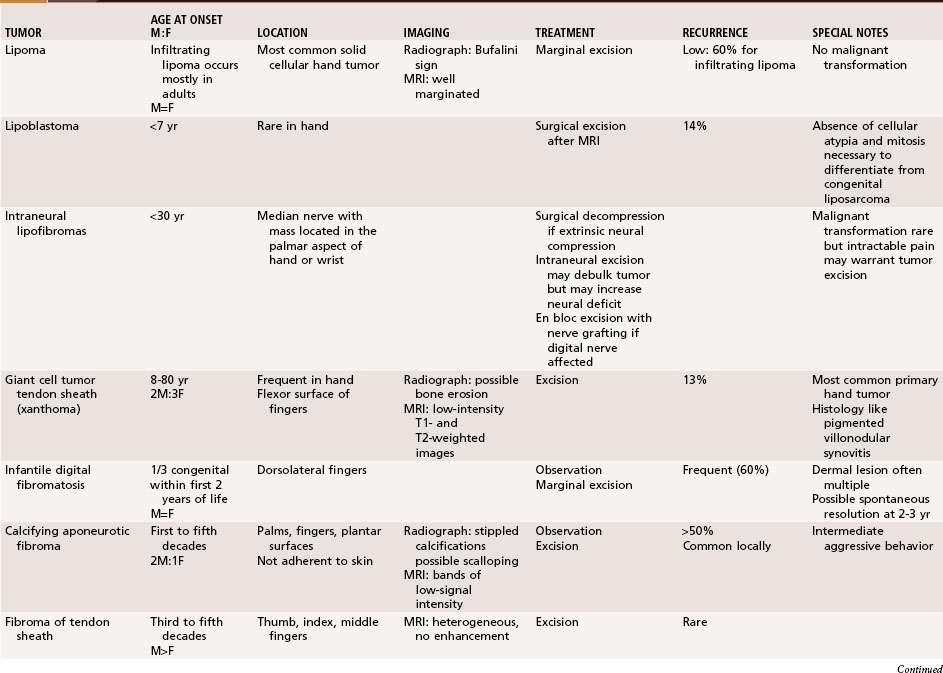
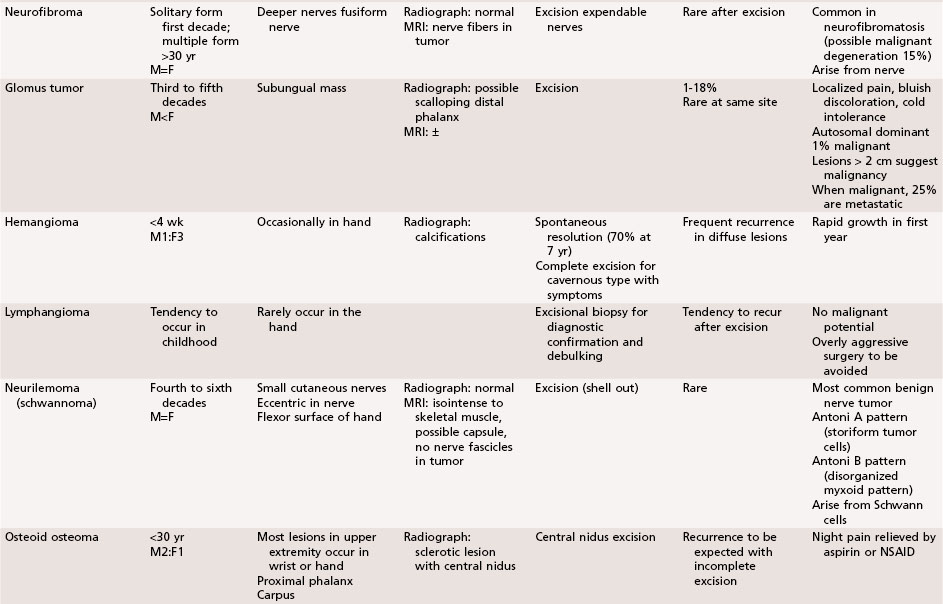
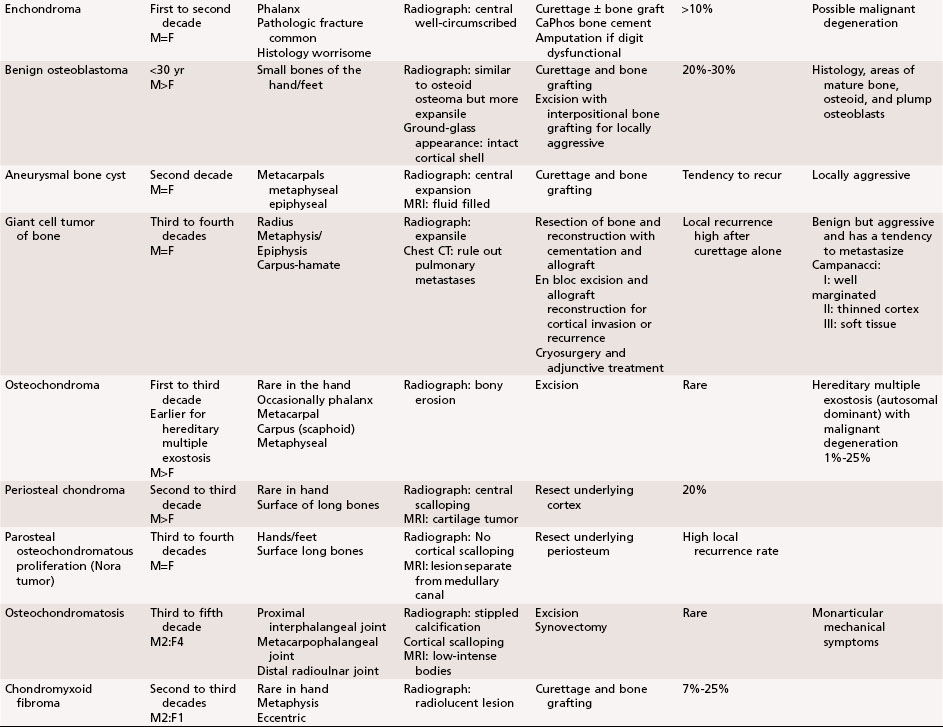
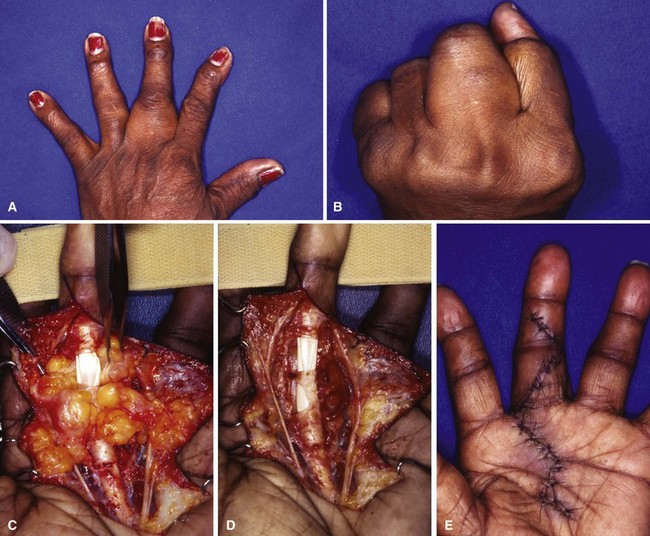
Intraneural Lipofibromas
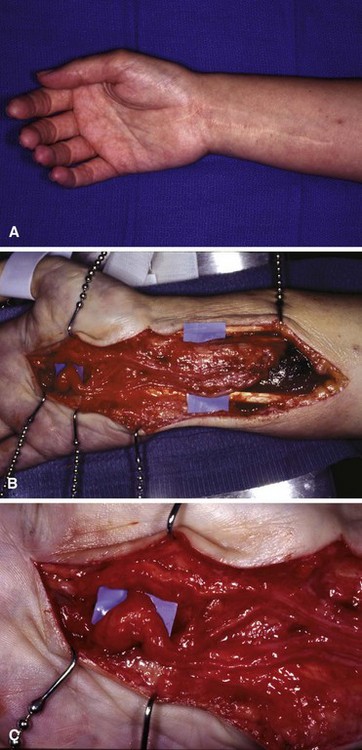
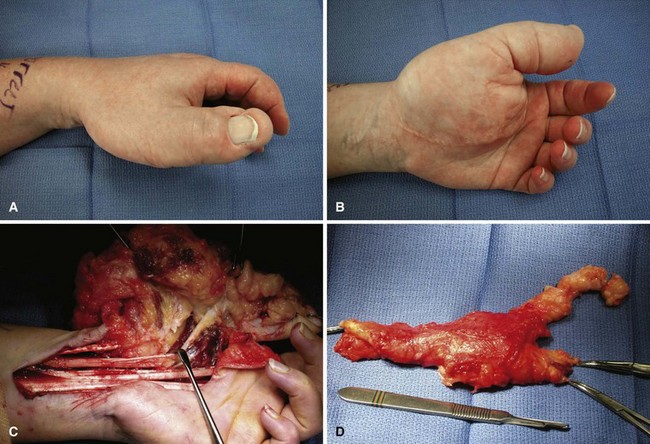
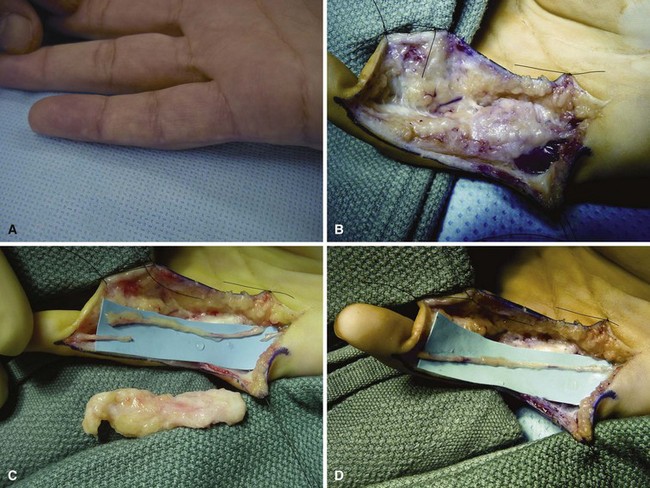
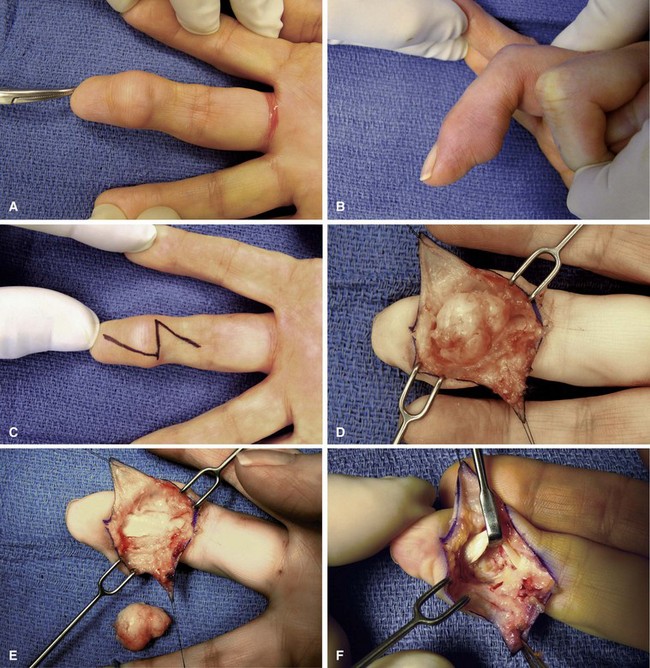
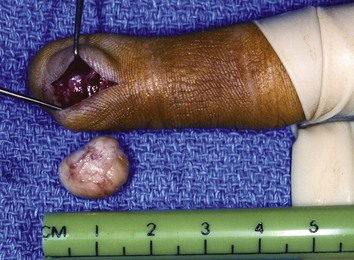
Intraneural lipofibromas are rare benign tumors that usually involve the median nerve. Patients present within the first 3 decades of life with a mass located in the palmar aspect of the hand or wrist, possibly with altered median nerve function (Fig. 77-2). Microscopically, fibroadipose tissue is seen infiltrating the epineurium, separating and compressing the fascicles. A conservative approach should be taken in treating these tumors. Incisional biopsy may be necessary for diagnosis. If extrinsic neural compression exists, surgical decompression should be performed by fasciotomy or carpal tunnel release. Intraneural excision may debulk the tumor, but this procedure is not recommended because intraneural fibrosis can increase the neural deficit. Malignant transformation is rare; however, intractable pain may result from some lesions and warrant tumor excision (Fig. 77-3). If a digital nerve is involved, en bloc excision with nerve grafting may be successful, although sensation would be altered (Fig. 77-4).
Giant Cell Tumor of the Tendon Sheath (xanthoma)
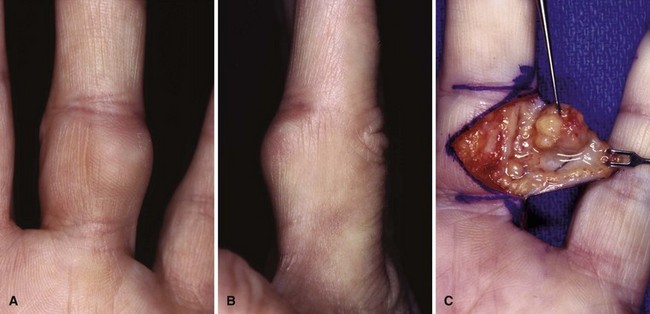
Giant cell tumors of tendon sheaths were first described in 1915 by Beekman, and some consider these to be the most common solid cellular hand tumors (Fig. 77-5). The reported age distribution is 8 to 80 years. These tumors occur in the hand more frequently than in any other part of the body, and they occur more often as firm lobulated masses on the volar lateral side of the index and middle fingers. Multiple xanthomas may be associated with hypercholesterolemia.
Benign Tumors of Fibrous Origin
Juvenile Aponeurotic Fibroma or Calcifying Aponeurotic Fibroma
First described by Keasbey in 1953, juvenile aponeurotic fibroma is a benign fibrous tumor typically appearing in the hands or wrists of children and young adults. It also has been called calcifying aponeurotic fibroma because in older patients calcification of the cartilaginous component may be present, a feature that distinguishes it from other benign tumors of fibrous origin. Clinically, it is a painless, solitary, and mobile mass less than 4 cm in diameter, usually involving the palm (Fig. 77-6). The tumor usually is on the volar side of the hand and is connected to peritendinous tissues and fascia. It has no gender predilection and no tendency to involve the ulnar side of the hand, as do Dupuytren nodules. Juvenile aponeurotic fibromas tend to develop close to tendons and are able to infiltrate surrounding muscle and fat. On radiographs, calcifications can be seen within the soft tissue mass. Because juvenile aponeurotic fibroma has a distinct tendency for local recurrence after marginal excision, especially in younger children (50%), a wide excision, preferably without sacrifice of function, is recommended.
Neurofibroma
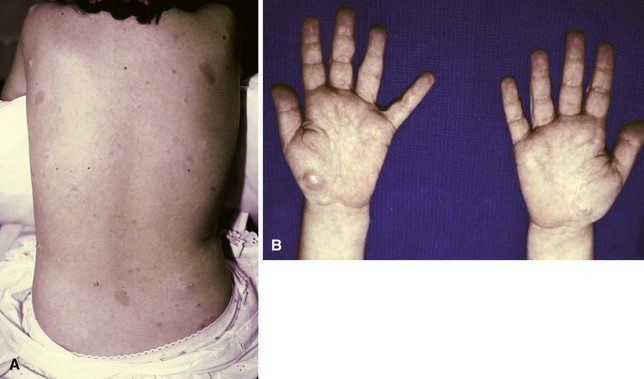
Neurofibromas rarely exist as solitary lesions, and most that occur as multiple lesions are associated with neurofibromatosis or von Recklinghausen disease. The solitary form usually occurs in the first decade of life, and the multiple form frequently manifests after 30 years of age. These lesions in the hand involve the more distal digital nerves (Fig. 77-7), where enlargement may produce grotesque finger angulation and gigantism. They usually are centrally located, nontender, more nodular, and nonencapsulated and may involve the skin, making them less mobile than schwannomas. The lesion is not resectable without sacrificing nerve elements because the nerve fibers intersperse in the tumor mass. Often these involve cutaneous nerves where proximal and distal to the lesion the nerve caliber appears normal. The solitary and multiple forms are histologically indistinguishable; both have a plexiform mass of irregular, thickened nerve fibers separated by increased endoneural matrix. Malignant transformation in neurofibromatosis has been reported to occur in 15% of patients, and complete excision is necessary for lesions that enlarge and become painful.
Glomus Tumor
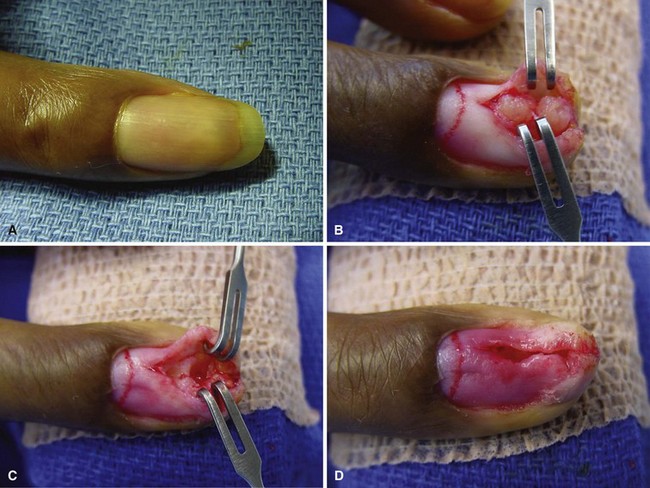
Glomus tumors usually are less than 1 cm in diameter, often being only a few millimeters in diameter, and may be visible through the overlying tissues as a deep red or purple discoloration. They occur more often in the hand (75%) than elsewhere and are located beneath the fingernail in 25% to 65% of patients (Fig. 77-8). Twenty-five percent are not subungual, however, and these may pose a difficult diagnostic problem. MRI and bone scans may prove helpful in diagnosing these tumors. Most of these tumors are benign; however, if the lesion exceeds 2.0 cm and histologic parameters suggest malignancy, then metastatic rates exceed 25%
Hemangioma
The following remarks are limited to the cavernous hemangioma and do not include the capillary superficial infantile hemangioma, which tends to involute by age 7 years. A cavernous hemangioma can be slightly to moderately tender and may enlarge a digit with distended venous sinuses. It produces a bluish color when it occurs close to the surface, and it forms a soft, collapsible mass. Calcifications often are visible on radiographs. Custom-fitted compression garments can be a useful conservative treatment. Radiation therapy is discouraged. Surgery is the treatment of choice for cavernous hemangiomas if symptoms justify it. The tumor can be so extensive that a staged procedure is required for its removal. Sometimes vessel ligation can assist a second-stage lesion excision. A rare coagulopathy, Kasabach-Merritt syndrome, can occur with lesions larger than 5 cm as a result of secondary platelet sequestration. Early treatment is indicated with this syndrome. With careful tourniquet control, blood partially fills the sinuses and outlines the extent of the tumor at the time of surgical excision. Complete excision usually is curative if the tumor is fairly well localized (Fig. 77-9); however, in diffuse lesions, persistence rather than recurrence is common. If complete excision is impossible, tumor debulking should be the emphasis of the procedure.
Neurilemoma (schwannoma)
Neurilemomas arise from Schwann cells or sheath cells (Fig. 77-10) and rarely are found in the hand despite being the most common solitary tumor of the peripheral nerves. Proliferation of the Schwann cells begins around one nerve fascicle and results in an eccentric or centrally located tumor. Two types of Schwann cells are present: hypercellular (Antoni A) cells, which are arranged in palisades, composed of plump and fusiform nuclei known as Verocay bodies, and hypocellular (Antoni B) cells, which are nonuniform cells dispersed in a myxomatous matrix. These tumors are not extremely tender and are more mobile at right angles to the course of the nerve than in line with the nerve. Neurilemomas frequently are misdiagnosed as ganglions and rarely are multifocal. With careful microsurgical technique, these tumors usually can be dissected from the surrounding nerve. Malignant degeneration is rare, and excision is curative. An alternative diagnosis or malignancy should be considered if the mass cannot be dissected free from the nerve trunk or is adherent to adjacent tissue. In such situations, incisional biopsy is indicated.
Osteoid Osteoma
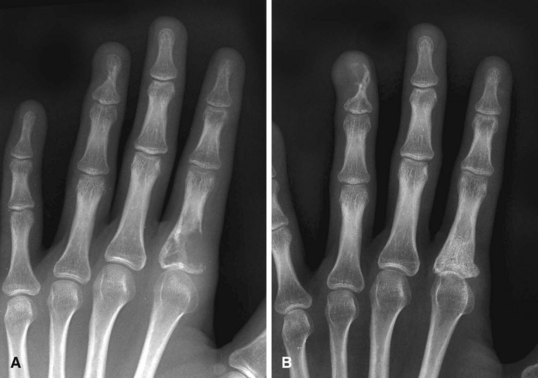
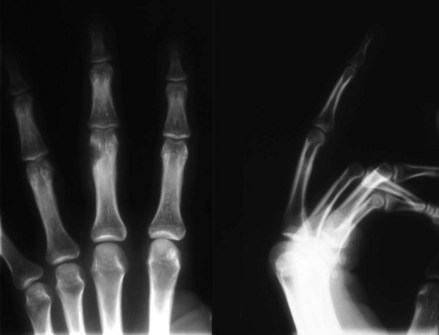
Osteoid osteomas are characterized by pain that gradually increases from mild to severe and usually is worse at night. Although dramatic pain relief can be obtained with aspirin, a small proportion of patients get no or only partial relief with nonsteroidal antiinflammatory medications. In one study, more than half of upper extremity osteoid osteomas occurred in the wrist and hand; the lesion occurred twice as often in men as in women, and the average age at diagnosis was 19 years (range 4 to 40 years). Some osteoid osteomas of the phalanges are painless, presumably because of the lack of nerve fibers trapped within the tumor. Generalized swelling of the involved part and tenderness to pressure are frequent findings. The carpus, especially the scaphoid, may be the site of involvement. The radiographic appearance depends on the area of bone involved. A small oval or round sclerotic nidus (Fig. 77-11) is surrounded first by an area of less dense bone, similar to a halo, and then by an area of sclerotic bone. Lesions in cortical bone or near the cortex may exhibit extreme sclerosis, requiring special imaging studies to reveal the nidus. A bone scan can be helpful in making the diagnosis. Treatment consists of creating a cortical window for complete removal of the nidus; recurrence may be expected if excision is incomplete.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


