Chapter 28 Soft Tissue Tumors
The evaluation of patients with musculoskeletal neoplasms was discussed in detail in Chapter 24, but certain points warrant repetition and elaboration. In contrast to bone tumors, the presence or absence of pain does not help to distinguish benign from malignant soft tissue tumors because most patients with soft tissue malignancies have minimal pain and consult a physician because of the presence of a mass. The mass typically is not invasive and grows in a centripetal fashion, pushing normal anatomical structures away. Careful physical examination of the mass, the involved part, and the lymph nodes draining the area is necessary. Standard radiographs provide little useful information about soft tissue tumors but may show phleboliths (hemangiomas), calcification (synovial sarcoma), or fat-density radiolucencies (lipomas). MRI may suggest a specific diagnosis in certain cases, such as lipoma, hemangioma, and pigmented villonodular synovitis. More often, MRI characteristics are nonspecific, but MRI is useful in evaluating the size and anatomical relationships of the tumor. Soft tissue sarcomas generally demonstrate low signal intensity on T1-weighted images and high signal intensity on T2-weighted images. CT also can be used for this purpose when a patient is unable to undergo MRI, but MRI is a superior imaging study in this circumstance. CT may help elucidate patterns of mineralization (i.e., calcification vs. ossification) found within the mass. Technetium bone scans usually do not provide significant benefit in the evaluation of the soft tissue mass but may be useful in select cases to assess local and distant bone involvement. For patients with malignant soft tissue tumors, a CT scan of the lungs should be obtained to look for metastases. Abdominal and pelvic CT scans are useful to detect retroperitoneal metastases in patients with myxoid liposarcomas and lymphatic spread in such lesions as synovial sarcoma, epithelioid sarcoma, and rhabdomyosarcoma. Biopsy often is required to establish the diagnosis and can be performed by core needle biopsy or by open incisional biopsy. Tissue sampling should be delayed until imaging studies are completed (biopsy will alter the study) and should be planned carefully according to the principles outlined in Chapter 24. Interpretation of the biopsy results may be facilitated by correlation with the clinical and imaging data. Staging can be performed using the Enneking system (see Table 24-1) or the American Joint Committee on Cancer system (see Table 24-2).
Benign Tumors and Tumor-Like Lesions (Table 28-1)
Fatty Tumors
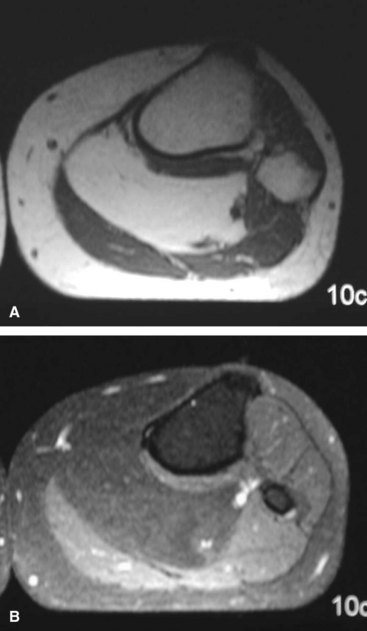
Lipomas are probably the most common benign tumors of connective tissue. They can occur at any age and in either sex but are probably more common in middle-aged women. These tumors usually develop subcutaneously but may involve the deeper structures. They occasionally affect the synovium (lipoma arborescens) and rarely the periosteum. Clinically, they are soft, circumscribed, movable masses that are painless and slow growing. A knee effusion is characteristically the presenting complaint in patients with lipoma arborescens. On radiographs, large masses appear as discrete radiolucent areas within soft tissue. MRI usually provides a definitive diagnosis because lipomas are uniformly bright on T1-weighted images and are dark on fat-suppressed sequences like the surrounding subcutaneous tissue (Fig. 28-1). Grossly, a lipoma is a well-encapsulated nodule of fat that may contain fibrous tissue. Microscopically, it is composed of mature fat cells with flattened nuclei, and mitotic activity is absent (Fig. 28-2). Some lipomas have a prominent vascular pattern and are appropriately referred to as angiolipomas. Focal areas with finely vacuolated cells of the brown fat type may be seen.
Nerve Sheath Tumors
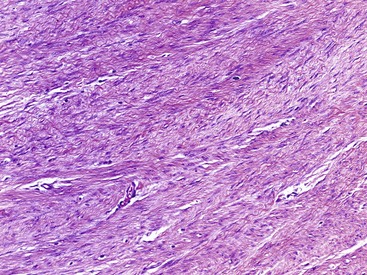
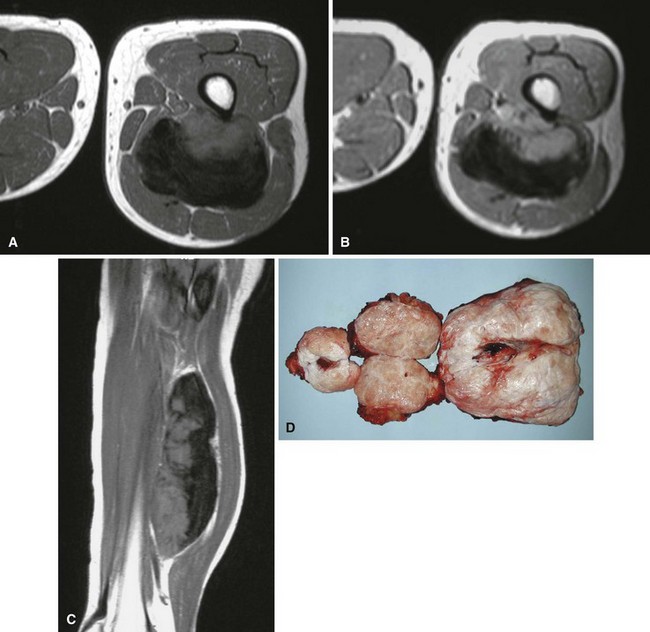
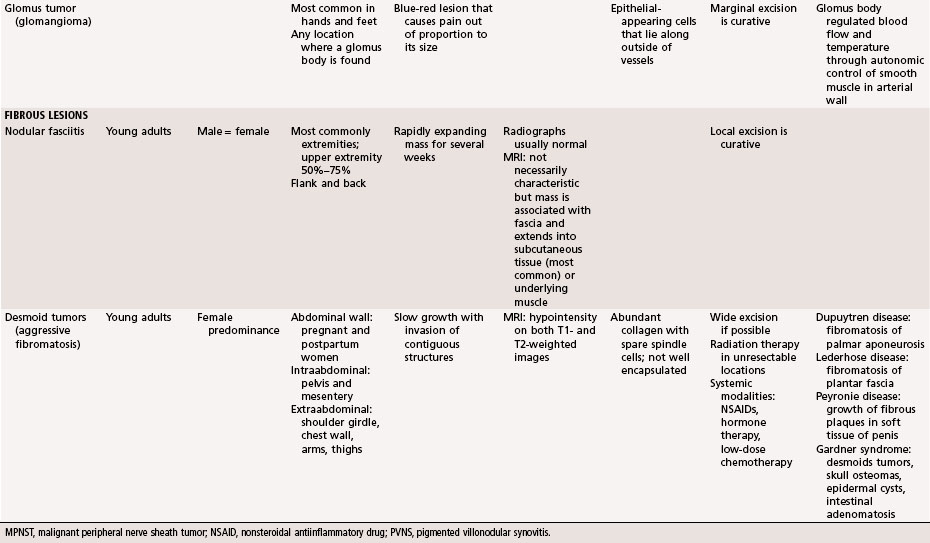
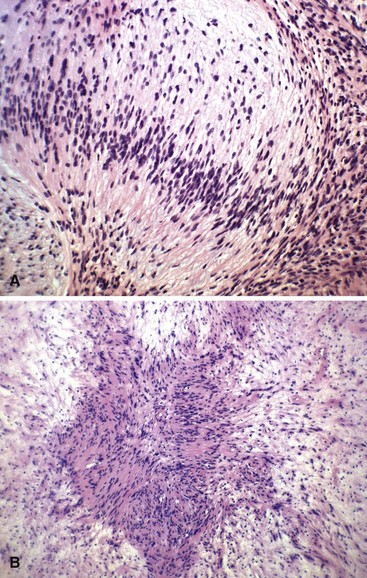
Neurilemmoma (schwannoma) typically is a solitary encapsulated lesion that may be cystic when it is 3 to 4 cm in diameter. Any nerve may be involved, but it usually involves one of the larger peripheral nerves (Fig. 28-3); the sacral plexus or the sciatic nerve may be involved within the pelvis. Patients may report a painless mass but sometimes complain of referred pain in the distribution of the involved nerve. A Tinel sign can be present on physical examination. These findings are extremely rare in association with other soft tissue masses except nerve sheath tumors. MRI often shows a fusiform mass along the course of a major peripheral nerve. The nerve may be visualized entering and exiting the mass on the coronal or sagittal MRI sequences. A “split fat sign” refers to a rim of fat that may be observed specifically on the T1-weighted MR image of schwannomas. However, MRI characteristics still remain nonspecific for this tumor. Microscopically, the tumor consists of two types of tissue: Antoni A and Antoni B. Antoni A tissue is more typical of the tumor and consists of compact collections of spindle cells that show marked palisading. Antoni B tissue is myxomatous and degenerative, and within it are cystic spaces and often thick-walled blood vessels. Verocay bodies are characteristically found in schwannomas and consist of an arrangement of two rows of palisading nuclei separated by fibrillary material (Fig. 28-4). Nuclear and cytoplasmic staining of the S-100 protein are not always required but help support the diagnosis. Often the lesion simply spreads the nerve fibers apart without anatomical or functional interruption, allowing the tumor to be removed by careful blunt dissection after a longitudinal incision in the perineurium. There may be little, if any, dysfunction of the nerve after surgery. Occasionally, the tumor may recur, but usually the lesion or recurrent lesion can be removed without sacrificing numerous nerve fibers. Excision that interrupts the continuity of the nerve should be avoided. Malignant degeneration has been described but rarely occurs.
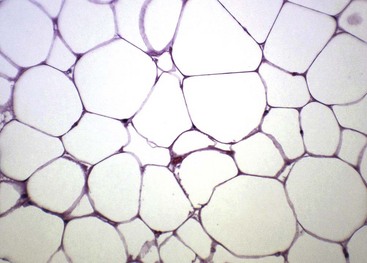
Neurofibroma is characterized by a much greater production of collagen than is neurilemmoma, and it, too, may occur as an isolated lesion (Fig. 28-5). It is much more likely than neurilemmoma to arise from a nerve branch too small to be identified, however. Neurofibromas also occur as a manifestation of von Recklinghausen disease (neurofibromatosis type 1), in which many such tumors may be found associated with café-au-lait spots and various other lesions. Most neurofibromas are found in individuals who do not have the NF1 marker (gene located at chromosome 17q11.2). Sometimes a neurofibroma occurs in which multiple fascicles of a peripheral nerve are involved; this is referred to as a “plexiform neurofibroma,” and excising it completely may be impossible without sacrificing the nerve and its associated function. A patient presenting with a plexiform neurofibroma without a known history of neurofibromatosis should be evaluated for neurofibromatosis type 1. Other manifestations of neurofibromatosis include hypertrophy of soft tissue, including the skin; hypertrophy of bone; scoliosis; bone cysts; and other abnormalities. Microscopically, neurofibromas have low cellularity with disorganized bundles of collagen. The nuclei of the spindle-shaped cells are wavy, and mitotic figures are rare (Fig. 28-6). Because neurofibromas are difficult to separate from the host nerve, axons may be observed histologically within the tumor cells. Even though the risk of malignant degeneration is low, increased pain or change in the size of the mass should serve as warning signs of a possible malignant peripheral nerve sheath tumor.
Synovial Lesions
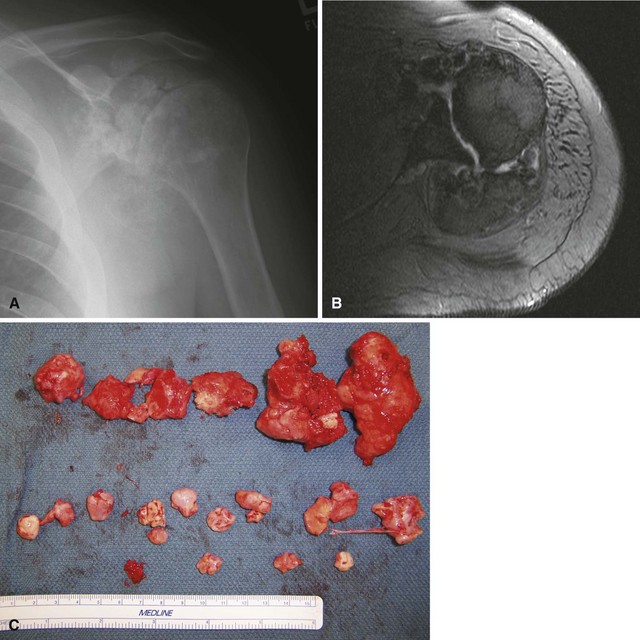
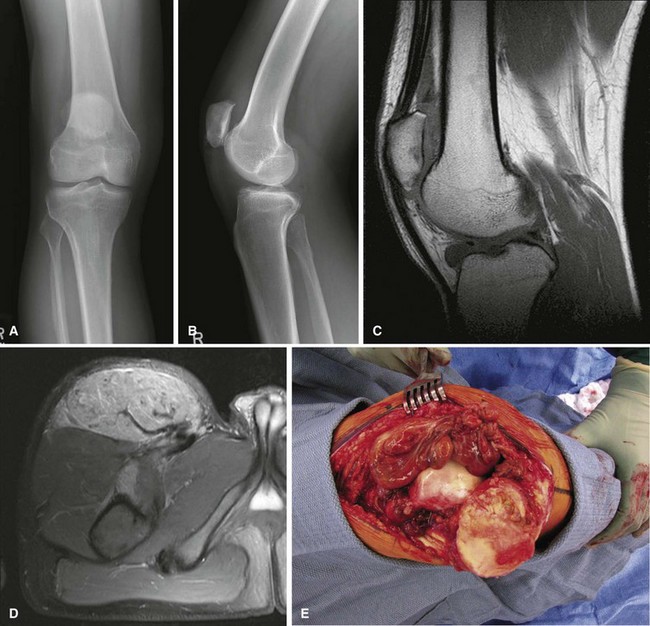
Synovial chondromatosis is a monarticular synovial proliferative disease in which cartilaginous or osteocartilaginous metaplasia occurs within the synovial membrane of joints, bursae, or tendon sheaths. The disease has been classified into three phases: (1) early, with synovial chondrometaplasia but no loose bodies; (2) transitional, with active synovial disease and loose bodies; and (3) late, with loose bodies but no synovial disease. Routine radiographs may show multiple loose bodies, and joint erosions may be present in late stages. MRI often is useful in establishing the diagnosis. Synovial chondromatosis is most common in the knee and hip, but almost any joint, bursa, or tendon sheath may be affected. Mechanical symptoms from the loose bodies may develop slowly over years and necessitate surgical intervention. Treatment consists of arthroscopic or open synovectomy and removal of the loose bodies (Fig. 28-7). Recurrence after surgery is common, and there are rare reports of malignant transformation to chondrosarcoma. Histologically, synovial chondromatosis consists of moderately cellular hyaline cartilage arranged in nodules within the synovium (Fig. 28-8). Binucleate cells may be observed, and cells are usually crowded and clumped within the nodule.
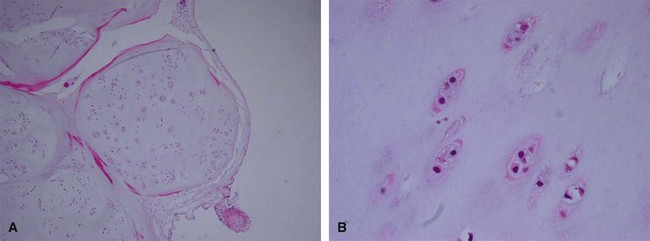
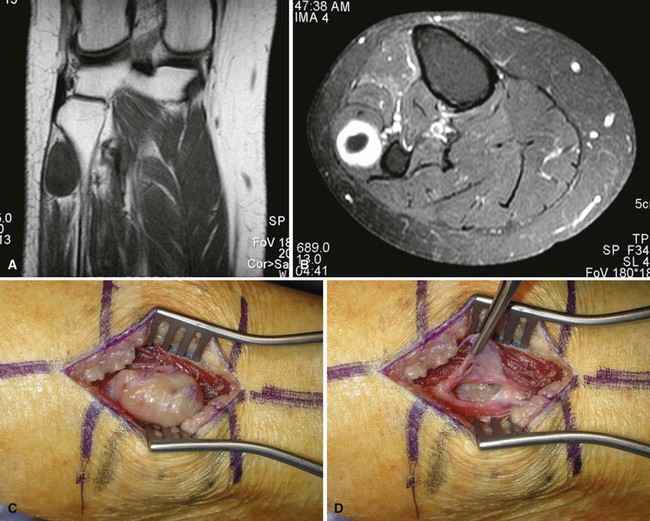
Giant cell tumor of tendon sheath is a relatively common benign tumor usually involving the tendon sheaths of adult fingers (Fig. 28-9). It first appears as a slowly enlarging but painless mass. Occasionally, a radiograph shows bony erosion of adjacent cortices. The histology is characteristic, with foam cells (histiocytes), fibrous tissue, giant cells, and hemosiderin deposition similar to that seen in pigmented villonodular synovitis (Fig. 28-10). Treatment is by marginal excision, which may prove technically difficult in larger lesions. Recurrences are frequent if the excision is incomplete.
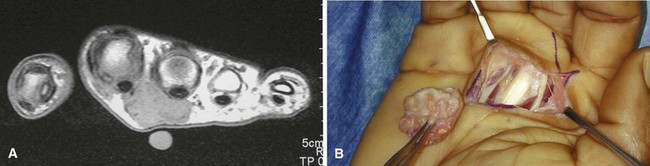
Pigmented villonodular synovitis may occur in a localized or diffuse form. The localized form is identical histologically to giant cell tumor of tendon sheath. The diffuse form also appears to be identical histologically to the localized form, but it involves the entire synovium. The diffuse form most commonly affects the knee, but the hip, ankle, shoulder, wrist, and other joints can be involved (Fig. 28-11). The patient usually presents with monarticular pain and swelling. Intraarticular lesions may cause mechanical symptoms, and a mass may be palpable. Aspiration of the joint characteristically reveals serosanguineous or blood-tinged fluid. Routine radiographs often are normal but may show bony erosion, especially if the hip is involved. MRI frequently is diagnostic, showing intraarticular masses that are dark on the T1-weighted and T2-weighted images. Additionally, the extent of the disease process may be further delineated with MRI. Recommended treatment for the localized form is marginal excision and for the diffuse form is total synovectomy. If there are significant secondary degenerative changes of the joint surfaces, arthroplasty should be strongly considered. Radiotherapy in the diffuse lesion may be justified if surgery fails to control the process.
Vascular Lesions
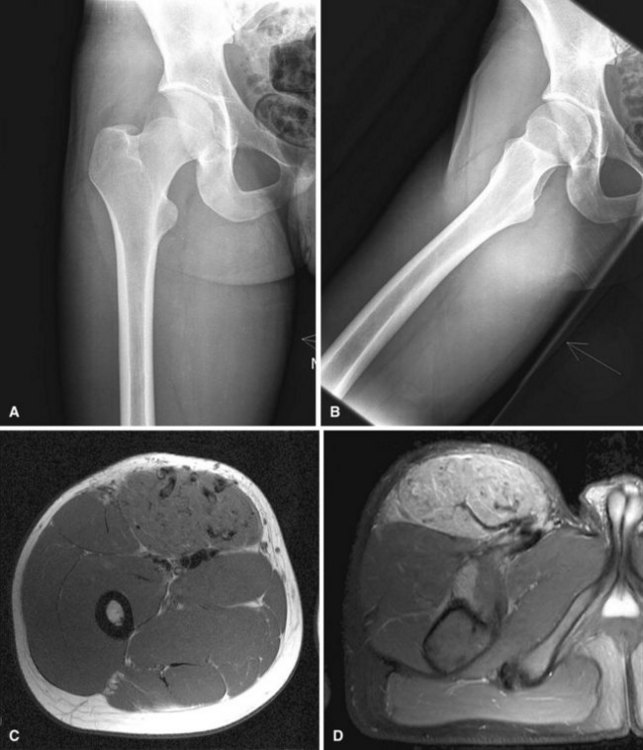
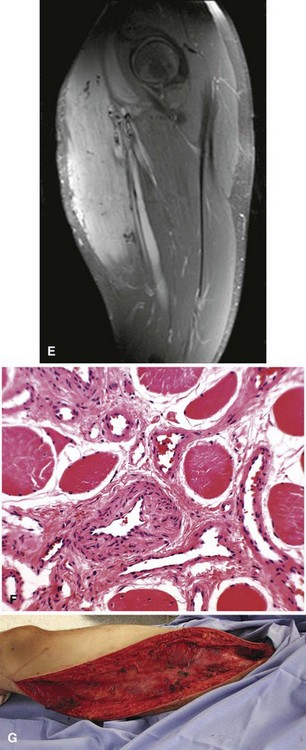
Capillary hemangiomas consist of a network of newly formed capillaries. Cavernous hemangiomas consist of widely dilated vessels or thicker vessels that may resemble veins; these occur quite often in the liver. Hemangiomas also occur deep in skeletal muscle and other soft tissues of the extremities and trunk (Fig. 28-12). Intramuscular hemangiomas can be painful. The pain often is associated with the increased blood flow that occurs during increased activity or when the limb is in a dependent position. Radiographs generally are normal but may demonstrate phleboliths or calcifications in the soft tissues. Hemangiomas characteristically have increased fat content as may be shown on MRI. The T2-weighted sequences will show increased signal secondary to the hyperemic vascular channels. Asymptomatic lesions can be observed. Multiple treatment options exist for painful lesions. Symptoms frequently can be controlled with compression hose. Surgical resection can be performed for small or well-defined lesions, although recurrence is common. Some intramuscular hemangiomas are infiltrative and extremely difficult to resect except by radical surgery. In these cases, treatment options include embolization or injection with sclerosing agents. Rarely, symptoms are refractory to all treatment options, in which case amputation could be considered.
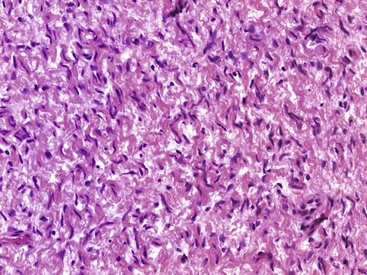
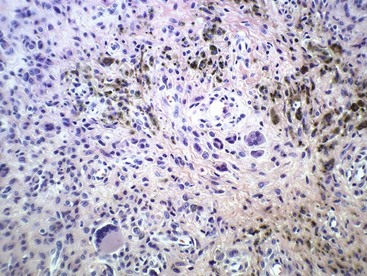
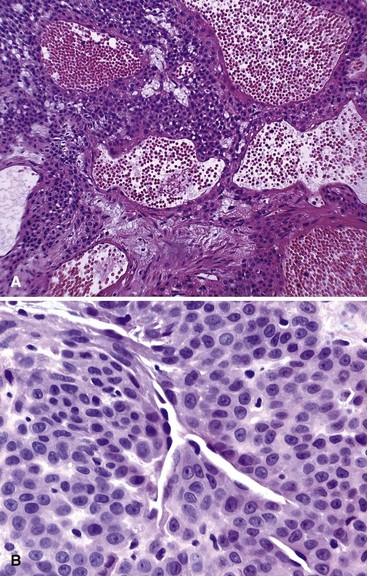
Glomus tumor, or glomangioma, is a rare but painful lesion. The skin and subcutaneous tissue of the hands and feet usually are affected, but a tumor may develop in any location in which a glomus body is found. A glomus body serves to control blood flow and temperature through the autonomic control of the smooth muscle of the arterial wall. The subungual area of the fingers is a characteristic site of involvement, especially in women. Exquisite point tenderness and hypersensitivity is invariably present. Because of this tumor’s rarity and small size, the diagnosis may not be readily apparent. When visible, the lesion usually appears as a small, bluish red discoloration of the skin, and 2% to 3% of glomus tumors are multiple. Multiple lesions are more common in men. Microscopically, this tumor consists of collections of round to oval uniform cells that appear epithelial and lie along the outside of abundant vessels (Fig. 28-13). Some smooth muscle also may be seen, and nonmyelinated nerve fibers may be shown by special stains. Glomus tumors are cured by marginal excision.
Fibrous Lesions
Several lesions have been described that microscopically consist of cellular tissue, often with bizarre cells and numerous mitotic figures, and that clinically have not behaved in a malignant fashion. One such lesion is nodular fasciitis (pseudosarcomatous fasciitis, subcutaneous pseudosarcomatous fibromatosis), which consists of bland proliferating immature fibroblasts in a myxoid stroma with a predominantly vascular pattern (Fig. 28-14). Typically, it is found in young adults, most frequently in the forearm, but it has been reported in a variety of ages and locations. The deep fascia usually is involved, with the lesion protruding into the subcutaneous tissue or less often into the underlying muscle. At its periphery, the lesion contains many capillaries and often inflammatory cells and resembles granulation tissue. Most lesions are relatively small (2 to 3 cm in diameter), but occasionally a larger one is found. Patients usually complain of a rapidly expanding mass that has been present for several weeks. Treatment is by marginal resection. Recurrence is rare.
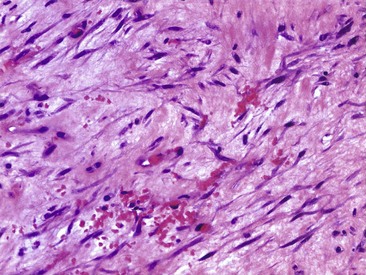
FIGURE 28-14 Nodular fasciitis. Photomicrograph (H&E) shows bland immature fibroblasts in a myxoid stroma.
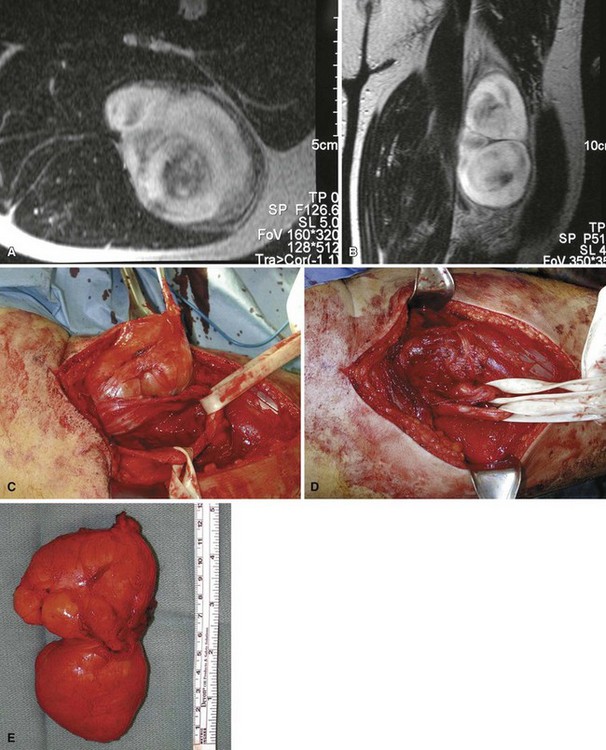
Desmoid tumors (aggressive fibromatosis) are locally aggressive lesions of connective tissue origin that infiltrate surrounding tissues and have a marked propensity for persistence or recurrence. Although most authorities regard this lesion as benign, it can be locally very aggressive. These lesions occur most frequently in the anterior abdominal wall of women who have borne children; lesions in other locations often are known as extraabdominal desmoids. Microscopically, they contain abundant collagen with uniform elongated spindle cells (Fig. 28-15). Atypia and mitoses are scant, and the mass is not well encapsulated, often infiltrating the surrounding host soft tissues. Grossly, they are dense, hard, rubbery, and grayish white. Extraabdominal desmoids occur most frequently in the shoulder girdle, arm, thigh, neck, pelvis, forearm, and popliteal fossa (Fig. 28-16). Superficial fibromatoses of the palmar aponeurosis (Dupuytren contracture) and plantar fascia (Lederhose disease) are commonly seen by the orthopaedist. The natural history of untreated lesions is unpredictable. Some patients experience a slow, relentless growth with invasion of contiguous structures. Spontaneous regression also has been reported. Metastases have been reported but are extremely rare. Most institutions currently recommend wide resection alone or marginal resection followed by adjuvant radiation therapy. For cases in which resection would involve unacceptable morbidity, radiation can be used alone. Subsequent resections for recurrence may prove arduous because of the difficulty in differentiating tumor from scar tissue. Other treatment options that have had reported success include systemic modalities such as tamoxifen, nonsteroidal antiinflammatory drugs, and low-dose cytotoxic agents.