Chapter 72 The Synovium
Normal and Pathologic Conditions*
Normal Synovium: Microanatomy and Function
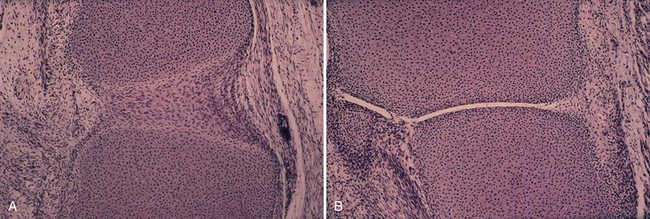
The synovium is formed when a primitive mesenchymal tissue cavitates, forming a recognizable joint space at approximately 8 weeks of embryonic life (Fig. 72-1). Mature synovium appears pale pink in color and architecturally covers all surfaces of the joint space, excluding the articular cartilage and most fibrocartilaginous structures. Only in abnormal conditions does the synovium encroach on the surface of the articular cartilage—a change classically seen in the reddish “pannus” or inflammatory synovial invasion of the articular cartilage in rheumatoid arthritis.
The synovial membrane, the most superficial layer of the synovium, lines the joint and forms the linings of tendon sheaths. Parietal and visceral synovium mimics their mesothelial-like counterparts in the thoracic and pericardial cavities. The extensive synovial-like lining cells of tendon sheaths and ligaments explain a host of reactive synovitis and painful clinical tenosynovitis, bursitis, and enthesopathy syndromes. Joint capsule fibrous connective tissue is thought to add to the joint’s mechanical strength. The term joint capsule refers to the fibrofatty neurovascular tissue that envelops the nonarticular cartilaginous tissue of the joint space.42
In pathologic states, synovial-like anatomic structures not related to a joint space (“synovial metaplasia”) are known to form in a variety of circumstances, including (1) postsurgical states; (2) failed prostheses, including breast implants29; (3) mechanical damage to connective tissue; and (4) experimental settings (injected air or oil in subcutaneous soft tissue).20
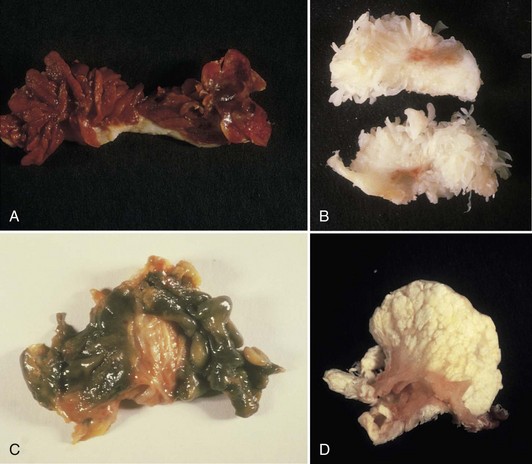
Normally, the synovium appears smooth and transparent, but it turns thick, dull, and opaque with pathologic change. With hemorrhage, it becomes obviously bloody, but in chronic hemarthrosis, it turns a reddish or rusty brown (Fig. 72-2) owing to hemosiderin deposition and the release of iron from red blood cells. In cases of severe bleeding, a dark purple or rusty color may be noted. A reddish purple or rusty synovium indicates a hemangioma or bleeding and may be seen in trauma, bleeding disorders such as hemophilia, von Willebrand’s disease, and pigmented villonodular synovitis (PVNS). In ochronosis (alkaptonuria), the synovium may appear a dull gray, whereas fibrocartilage and articular cartilage are discolored black. Darkening or blackening also may be seen with extensive release of metallic debris. White foci in the synovium usually indicate gout (urate deposition), pseudogout (calcium pyrophosphate deposition [CPPD]), or soft tissue calcifications (deposits caused by trauma or calcinosis syndromes). Cement debris also may lead to pallor. A yellow color ensues with xanthoma cell accumulation, which may be a predominant feature of PVNS or xanthomatous disease.
Microanatomic Structure
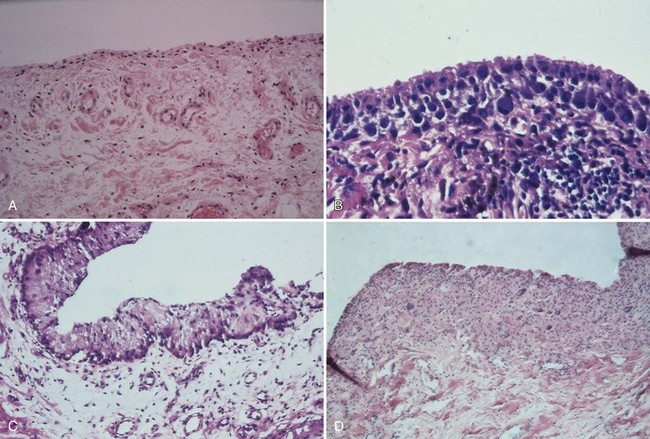
The synovium consists of a thin layer of synovial cells or synoviocytes, the intimal layer, above a richly fibrovascular zone, the subintimal layer, which contains arterioles, fat, and other connective tissue cells, such as fibroblasts, histiocytes, and occasionally mast cells (Fig. 72-3). The intimal layer of lining cells is usually one to two cells thick with no discernible differences on light microscopy. The loose connective tissue subintimal zone layer becomes gradually more fibrous at capsular insertions.
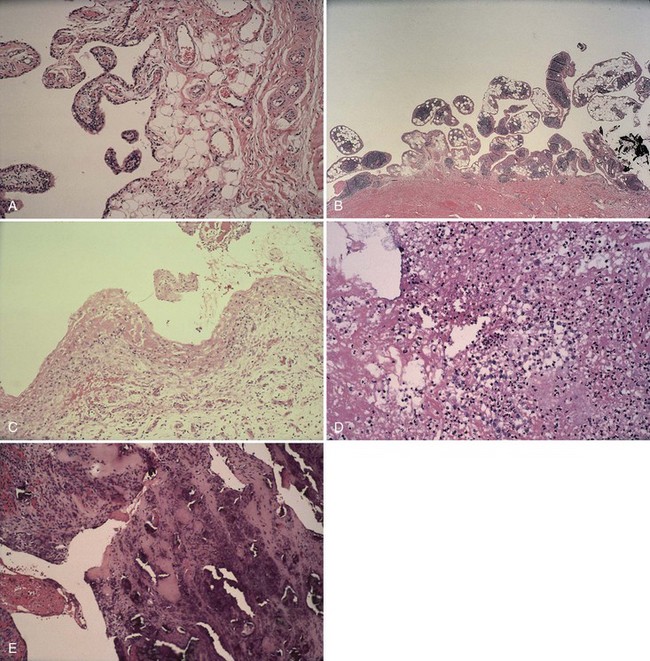
The villous appearance of synovium is not abnormal but is rather nonspecific and may be seen in a broad range of conditions (Fig. 72-4). In general, traumatic synovitis and degenerative joint disease (DJD) (osteoarthritis) are attended by edematous change and mild villous hypertrophy. Inflammatory arthritis (classically rheumatoid arthritis) shows a dramatically reddish hyperplastic synovium with fibrinous exudation characterized by abundant tan fibrinous loose bodies, called rice bodies, and marked lymphoplasmacytic synovitis. In septic arthritis, leukocytes are seen in tissue. Neuropathic joints and rapidly destructive joint processes are characterized by bone and cartilage debris.
Ultrastructure
The intimal zone consists of an admixture of cell types often conveniently classified as cells exhibiting macrophage function (synovial A cells) and cells more synthesizing in function (synovial B cells) (Fig. 72-5). Electron microscopy studies and immunophenotyping studies further characterize these cells. Ultrastructural studies show abundant mitochondria, Golgi apparatus, vacuoles, lysosomes, phagosomes, vesicles, and surface undulations (characteristics suited to macrophage activity) in type A cells, and rough endoplasmic reticulum, free ribosomes, and smoother cytoplasmic profiles (characteristics suited to synthetic activity) in type B. As might be expected, synoviocytes may be “intermediate” in nature, featuring organelle functions of types A and B. Some evidence supports the existence of other cells, such as antigen-type cells (HLA-DR, IA-like).
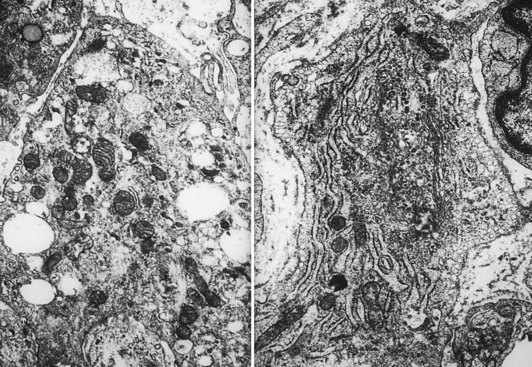
Arbitrarily designated type 1 (synovial “A” cells) is a group of cells that seem to be related to mononuclear phagocytes based on their expression of antigen and derivation from cells of the monocyte macrophage cell lineage.22 These cells, which exhibit phagocytosis and contain macrophage markers, abundant HLA-DR IA antigen, and Fc receptors, constitute approximately one third of the synovial cell population.
Cell types as defined by immunologic studies are consistent with previous morphologic observations. Type 1 cells expressing the monocyte macrophage antigens are similar to the cells historically described as characterized by phagocytosis type A and type C cells. Type 1 cells are most likely mononuclear phagocytes of bone marrow origin, as ascertained by their antigenic marking with monocyte and IA antigen. Modifications in antigen expression suggest that they act more like tissue macrophages in the joint. The historically designated “type B” synovial cell that produces glycosaminoglycans is most likely of the type 3 cell variety, probably mesenchymal in origin. These cells lack IA antigens and most of the differentiated antigens of monocyte macrophage lineage.11
Although synovial cells lack desmosomes or tight junctions, characteristic of epithelial tissue, the complexity of this cell structure is evident in the changes seen in various pathologic states. Hyperplasia may be limited to a mild increase in intimal cell number, or dramatic change, including large, bizarre cells, such as Grimley-Sokoloff giant cells or even striking mucin-producing cells, may occur. In this latter condition (mucinous hypertrophy of the synovium), the copious amount of material secreted testifies to the potential capacity of this membrane (see Fig. 72-3C). More recently, multipotential mesenchymal stem cells were developed from the adult human synovial membrane and were inducible into chrondrogenesis, myogenesis, osteogenesis, and adipogenesis.16
Synovial Fluid
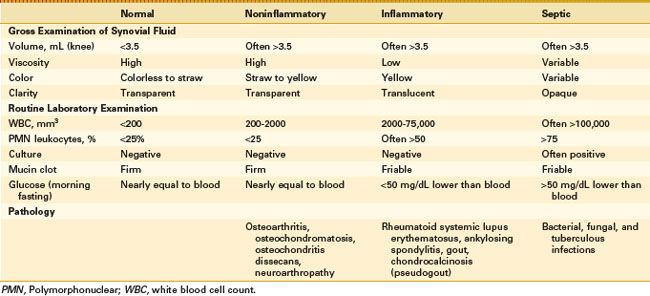
Synovial fluid analysis may be grouped for general classification of types of conditions producing synovial fluid (Table 72-1 and Fig. 72-6).73 Noninflammatory conditions, such as DJD, trauma-induced arthritis, osteochondritis dissecans, and neuropathic arthropathies, or primary synovial metaplastic conditions, such as synovial chondromatosis, are characterized by a yellowish synovial fluid with high viscosity, low leukocyte counts, less than 10 mg/dL of glucose (i.e., below serum level), and low protein. Cultures are negative. This noninflammatory or class I type of synovial fluid group is distinct from inflammatory or crystal-induced arthropathies, which are characterized by a synovial fluid with low viscosity, a yellow or whitish color, and a variable leukocyte count with a greater than 50% neutrophil population. Glucose usually is elevated to more than 25 mg/dL, and protein is elevated to more than 3 g/dL.
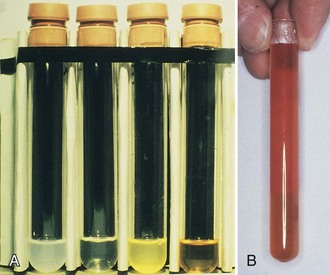
Septic arthritis usually is characterized by a yellowish green fluid of low viscosity with an elevated leukocyte count and greater than 75% neutrophils. Glucose is usually greater than 25 mg/dL with increased protein levels (Figs. 72-7 and 72-8). A wide range of conditions can induce a hemorrhagic arthropathy, including idiopathic hemosideric synovitis, trauma-induced arthritis, hemophilic arthropathy, and PVNS as a secondary change. These synovial disorders are characterized by a reddish brown color with decreased viscosity and a moderate elevation in white blood cell count with greater than 25% neutrophils. Glucose is in the normal range, usually 0 to 10 mg below serum level. Protein may be greater than 3 g/dL.
Iron-Related Changes
Iron in tissue removed during orthopedic or related conditions usually is seen in the form of hemosiderin, typically identified as granular brown pigments in an intracytoplasmic localization. The presence of hemosiderin can be noted in a broad variety of conditions (Box 72-1).73 Traumatic hemarthrosis is seen in association with soft tissue injuries and fractures, including secondary fractures associated with other pathologic states. Iron can be a contributing factor to the pathophysiology of disorders such as hemophilic arthropathy and transfusional hemosiderosis of thalassemia. Deposition at the mineralization front, causing osteoporosis or even osteomalacia, can be seen in primary or secondary hemochromatosis. The term pigmented in PVNS refers to the brown pigmentation caused by coincidental iron deposition in synovial tissue surrounding the proliferating nodules of an essentially fibrous tumor. A yellow color may be seen as a result of the abundant presence of foamy histiocytes.
Acute hemarthrosis of the knee in athletes may result from a wide range of injuries to the meniscus and the cruciate ligaments, fracture of the bone, and tear of synovial tissue.52,71 In chronic hemarthrosis, iron accumulates in the synovium. Histopathologic localization includes the synovial intimal cells and the histiocytic cells of the subintimal zone. Grossly, the synovium may develop a “rusty” appearance (see Fig. 72-3).
Experimental evidence suggests that iron adversely affects synovial function. Chronic hemarthrosis may increase the synthetic function of the otherwise macrophagic synovial type A cell. Hemosideric synovitis without hemophilia is well described clinically.23 Hemophilia represents this situation in a clinical extreme.
Findings on magnetic resonance imaging (MRI) in iron or hemosiderin accumulation are complex. In general, hemosiderin has a so-called paramagnetic or ferromagnetic property, causing a signal dropout37 in most cases.
Hemosideric Synovitis
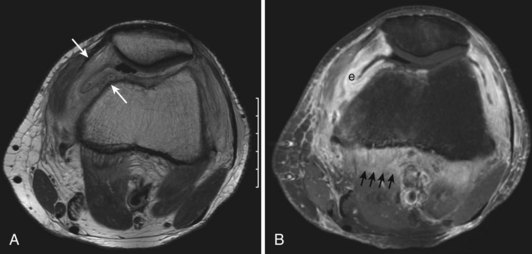
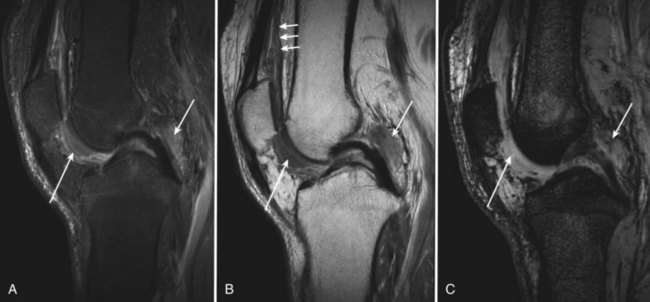
With intra-articular hemorrhage, one may detect fluid–fluid levels depending on the extent and duration of the bleed (Fig. 72-9). In hemorrhages that occur within the synovium, which is a highly vascularized tissue, red blood cells eventually disintegrate, and phagocytosis is begun by histiocytes. Eventually, the hemoglobin is broken down and processed into hemosiderin. Hemosiderin characteristically is seen as a brown-pigmented granular substance in cells of the synovium. Hemosiderin may occur as minute granules or may accumulate into globules 25 µm in diameter. The intracellular deposition of hemosiderin is observed mostly in macrophages or histiocytes, but also may be seen engulfed in various trauma-associated conditions by hypertrophied synovial lining cells. In joints subjected to hemorrhage, hemosiderin may be noted intracellularly or extracellularly. Cells within different compartments or layers of the synovium may be affected, ranging from synovial lining cells to subsynovial proliferating connective tissue cells. The gross appearance of the joint with chronic hemarthrosis includes a rusty pigmentation, although more acute bleeding may be demonstrable as blackish green discoloration.
With bleeding into the joint, two pathways may lead to damage. Under one mechanism, the red blood cells may break down, causing macrophage activation. Ensuing inflammation may lead to destructive changes in and of themselves, as seen in rheumatoid arthritis. Intracellular hemosiderin precipitates the release of leukocyte-derived and synovium-derived chondrocytic enzymes.6 In excessive cases, iron deposition in organs such as the meniscus may be severe enough to cause mechanical dysfunction and degenerative changes. In a second pathway, bleeding may lead directly to synovial proliferation and the release of an uncontrollable cascade of destructive substances such as proteases.
Hemophilia
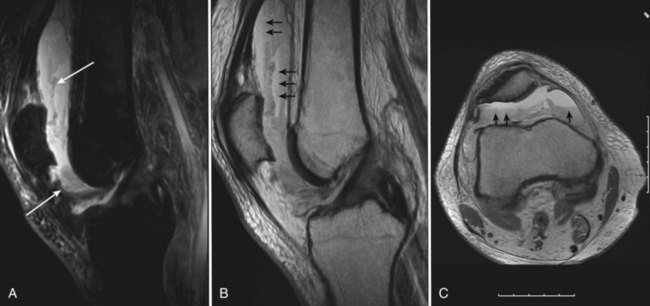
Adverse effects on synovium in patients with chronic hemorrhage have been studied most extensively in the joint changes of hemophilic arthropathy of the knee. In tissue removed from hemophiliac patients, the synovium shows marked villous hypertrophy with extensive hypertrophy and hyperplasia of synovial lining cells and with subsynovial connective tissue components with abundant intracytoplasmic hemosiderin granule accumulation (Fig. 72-10). In tissue cultures of synovium in hemophilia, pigment-laden fibroblast cells have been shown to proliferate, and explants of synovial cells have been shown to secrete large quantities of latent collagenase and neutral proteinases, establishing the destructive potential of synovitis in hemophilia. Synovium incited by hemophilia produces enzymes and may do so without coincident inflammation; the latter is a significant component of the destructive arthropathy seen in rheumatoid arthritis.
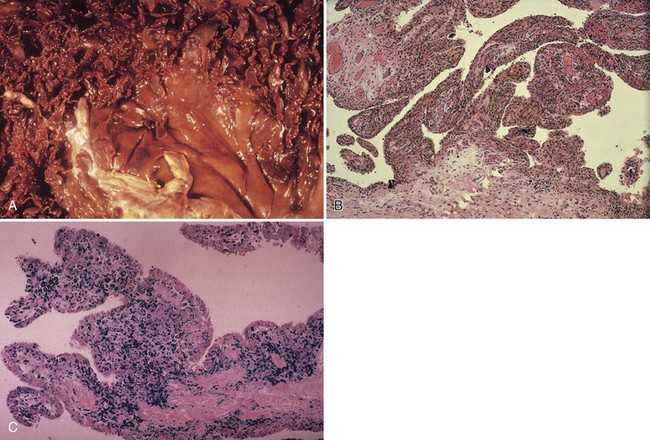
Although it is less visible, iron is known to accumulate in cartilage as well. Extensive hemosiderin deposition leading to grossly visible brown menisci has been observed clinically.6 In the study of hemophilic joints, iron has been localized to superficial chondrocytes, suggesting chondrocytic phagocytic uptake triggering a degradative enzyme release similar to that described in synovial cells. Iron has been localized histochemically to the tidemark in lupus and in hemochromatosis. Hemophilia A and hemophilia B are inherited X-linked deficiencies of factor VIII and factor X.
Hemophilia A is the most common hereditary coagulation disorder, affecting 1 per 5000 live male births as the result of deficiency, absence, or malfunction of coagulation factor VIII.65 Roughly one third of patients have no history of this disease. In normal patients, a very low concentration of factor VIII (0.2 mg/mL plasma) is sufficient for adequate coagulation. Bleeding leading to clinically perceptible illness requires a reduction of at least 75%.
Hemophilia A is a clinically heterogeneous disorder ranging from mild disease (1% to 4% deficiency) to severe disease. Patients with mild or moderate disease may not be recognized unless a significant traumatic event precipitates abnormal bleeding. Excessive bleeding during surgery may be the first clue. Therapy is centered on replacement of factor VIII.25 Replacement traditionally was done with shotgun frozen plasma, and cryoprecipitate became available in the 1960s. More recently, purified concentrates have been used. Current treatment consists of regularly scheduled replacement of deficient factor with recombinant proteins synthesized in tissue culture. In general, 1 U of factor VIII increases plasma activity by 0.024 U/mL. Because 0.3 U/mL usually is needed to treat a mild bleeding episode, clinical goals should strive for more than this amount. Therapy has significantly improved morbidity.
Radiographically, joint space narrowing, synovial proliferation, loss of articular cartilage, cystic remodeling of bone, and hemophilic pseudotumors characterize the illness (Fig. 72-11). Strict adherence to maintenance of factor VIII levels to prevent spontaneous hemorrhage has been associated with significantly decreased morbidity and has halted radiographic progression of disease.
Surgical joint reconstruction in hemophiliac patients has been attempted in an effort to preserve function, but radioactive synoviorthesis and arthroscopic synovectomy have been used.6 Total knee arthroplasty performed for hemophilic arthropathy has a high risk of failure as a result of infection, which develops late and often is caused by Staphylococcus epidermidis.59
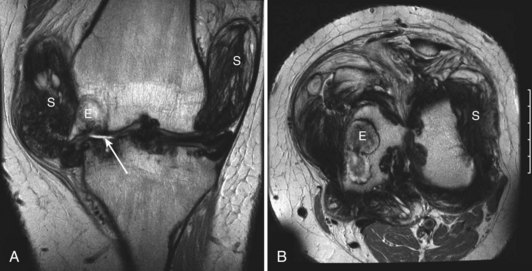
Intra-articular, intrabursal, and soft tissue bleeding in hemophilia may result in painless masses that clinically and radiographically mimic a tumor, called pseudotumors of hemophilia (Fig. 72-12).24 These masses consist of spongy coagula of partially clotted blood encapsulated by thick fibrous membranes. Complications of these so-called hemophilic pseudotumors include muscle and bone damage, infection, and neuropathies. Surgical removal is not undertaken without danger. Pseudotumors occur in 1% to 2% of hemophiliac individuals,6 mostly in the lower extremity and pelvis. Bleeding in the vicinity of the periosteum has been implicated in peculiar juxtaosseous changes, among them cystlike bone changes and large soft tissue masses that erode bone. MRI studies indicate low signal on T1-weighted imaging and high signal on T2-weighted imaging.
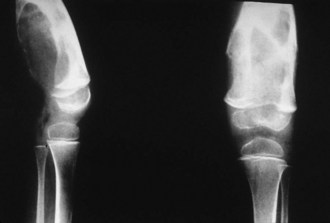
Figure 72-12 Radiograph of hemophilic pseudotumor.
(From Vigorita VJ: The synovium. In Vigorita VJ [ed]: Orthopaedic pathology, Philadelphia, 2008, Lippincott Williams & Wilkins.)
Before 1970, hemophilia A was associated with significant severe disability and death at a young age. Median life expectancy grew significantly throughout the 20th century, however, from 11.4 years to 68 years in carefully monitored populations.47 Transfusion of blood products, including factor VIII concentrates, has led to the well-recognized modern complication of hemophilia—transfusion-related acquired immunodeficiency syndrome (AIDS). Life expectancy now has reversed after considerable gains. Large numbers of hemophiliac individuals developed serologically detectable antibodies to human immunodeficiency virus (HIV) beginning around 1979. Approximately two thirds of HIV-positive hemophiliac patients have eventually died of AIDS. Risk to the surgeon is minimal if proper precautions are taken.30 More recent therapy has used factor VIII concentrates exposed to vigorous virus-killing heat treatment or solvent cleaning. Genetically engineered products now available are being tested for complications. Expense is a significant consideration.
Hemochromatosis
The cause of secondary hemochromatosis is ineffective erythropoiesis, as seen in β-thalassemia and sideroblastic or aplastic anemia, for which treatment may include repeated transfusions. Secondary hemochromatosis also may be caused by chronic liver disease, and in certain populations, such as within sub-Saharan Africa, hemochromatosis has been attributed to increased intake of dietary iron, such as from large amounts of iron in beer brewed in steel drums. Individuals at greatest risk probably have a genetic predisposition.27
The deposition of iron in bone, particularly at the osteoblast–osteoid interface or at the mineralization front, or as hemosiderosis within hematopoietic cells of the marrow, is a relatively common sequela to primary and transfusional hemosiderosis. Although the primary organs involved are the heart and the liver with eventual development of cirrhosis and hepatocellular carcinoma and heart failure, endocrine organs are particularly involved. Joint pain in the clinical presentation of arthritis has been noted in 11% of patients carefully analyzed clinically; bone pain is a less frequent presentation.3
Lead Synovitis
The signs and symptoms of lead poisoning are subtle, and recognition of this entity may be difficult. Plumbism is caused most commonly by ingestion of lead-based paint by children; by occupational exposure, such as painting, lead mining, and working in battery factories; and by consumption of contaminated beverages, such as moonshine.9
A prototype of lead injury to the joint is the effect of retained bullet fragments.8 Lead poisoning secondary to retained projectiles is rare (Fig. 72-13) but has been reported since the ancient Roman wars. Lead toxicity may cause convulsions, somnolence, mania, delirium tremens, coma, neuritis, nausea, vomiting, abdominal cramps, anorexia, weight loss, renal insufficiency, general malaise, and death. Orthopedic complications include bone cysts, localized arthropathy, pseudarthrosis, and gouty arthritis. Serum levels of lead of 920 µg/dL (44.40 mm/L) have been reported after retention of lead projectiles. The time between injury and onset of symptoms has ranged from 2 days to 40 years, although patients may be asymptomatic for long periods.
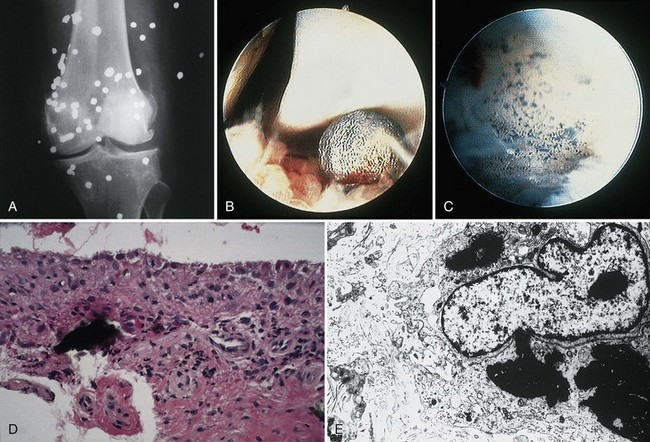
Classic radiographic findings are those of intra-articular speckled lead deposits, joint space narrowing, and preserved bone density.21
Related posts:
 Cemented Total Knee Arthroplasty: The Gold Standard
Cemented Total Knee Arthroplasty: The Gold Standard
 Management of Extra-articular Deformity in Total Knee Arthroplasty With Navigation
Management of Extra-articular Deformity in Total Knee Arthroplasty With Navigation
 Patellar Instability
Patellar Instability
 Posterior Cruciate Ligament Reconstruction: Posterior Inlay Technique
Posterior Cruciate Ligament Reconstruction: Posterior Inlay Technique
 Anterior Cruciate Ligament Reconstruction With Central Quadriceps Free Tendon Graft
Anterior Cruciate Ligament Reconstruction With Central Quadriceps Free Tendon Graft
 Osteochondritis Dissecans
Osteochondritis Dissecans
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


