Figure 3.1 Comparison of the response of the exercise tidal flow volume in a person with expiratory flow limitation (EFL) (solid lines), compared with that predicted for someone with normal lungs (dashed lines). Note that in the presence of EFL there is encroachment upon the inspiratory capacity in order to increase minute ventilation (flow volume loop shifts to the left and end-expiratory lung volume (EELV) increases). The person with normal lungs (dashed lines) is able to increase minute ventilation by utilizing both their inspiratory and expiratory reserve volumes (EELV decreases). The inset illustrates the pressure–volume relationship of the inspiratory muscles showing that, as lung volume increases from residual volume (RV) towards total lung capacity (TLC) (as occurs in hyperinflation) the inspiratory muscles become weaker.
Hyperinflation has also been shown to impair respiratory muscle blood flow in a dog model (Kawagoe et al, 1994); in this study, despite an almost two-fold increase in the work of breathing, diaphragm blood flow remained unchanged and accessory muscle blood flow fell during acute hyperinflation. It is not clear whether hyperinflation exerts the same effect in human beings with COPD, but impaired accessory muscle perfusion in the face of an increased demand for muscle work would predispose these muscles to fatigue and / or accumulation of metabolic by-products (see section ‘Respiratory muscle involvement in exercise limitation’).
Thus, COPD-induced changes in respiratory mechanics exert a very potent influence upon dyspnoea because they affect both the demand for inspiratory pressure generation and the capacity of the inspiratory muscles to generate sufficient pressure to meet that demand (see Fig. 3.1). Both phenomena increase the requirement for inspiratory motor drive and intensify dyspnoea (O’Donnell, 2001). However, the inspiratory muscle dysfunction of COPD is not confined to the functional (secondary) weakening precipitated by hyperinflation (Similowski et al, 1991; Polkey et al, 1996). There is also primary dysfunction due to abnormalities within the muscle tissue itself, which lead to declines in strength and endurance (Levine et al, 2003; Barreiro et al, 2005; Ottenheijm et al, 2005). This deterioration of muscle may be in part due to disuse (sedentary lifestyles), but is more likely to be the result of oxidative stress (Barreiro et al, 2005) resulting from the systemic manifestations of COPD, including the chronic inflammatory state. Furthermore, malnutrition causes generalized muscle weakness, which may exacerbate disease-specific respiratory muscle weakness (Decramer, 2001). Finally, the use of oral corticosteroids has been shown to have a myopathic influence upon the respiratory muscles of patients without respiratory disease, who show significant reductions in strength (~ 30%) and endurance (~ 50%) over the treatment period (Weiner et al, 1993; Weiner et al, 1995). Although these changes show some reversal following cessation of corticosteroid treatment, function may take as long as 6 months to normalize (Weiner et al, 1993). Since primary and secondary dysfunction coexist, there is a significant impairment in the capacity of the inspiratory muscles to deliver changes in intrathoracic pressure and tidal volume. Indeed, disease severity correlates negatively with respiratory muscle function (Terzano et al, 2008). Furthermore, hyperinflation leads to changes in chest wall geometry, inducing functional weakening of the accessory inspiratory muscles, which also contributes to a global reduction in the ability of the respiratory pump to generate inspiratory pressure (De Troyer & Wilson, 2009).
Much has been made in recent years of the adaptations that occur within the inspiratory muscles in response to the mechanical changes and increased physical demands described above. The chronically hyperinflated, flattened state of the diaphragm in COPD appears to lead to shortening of the total diaphragm length by around 15% to 25%, depending upon whether this is assessed at functional residual capacity FRC or residual volume (RV) respectively (McKenzie et al, 2009). This adaptation reduces the ability of the diaphragm to shorten during contraction, and thus limits its ability to generate inspiratory flow. However, the adaptations in diaphragm geometry and length appear to have some functional benefits in terms of maintaining its ability to deliver volume excursion, as well as its pressure-generating capacity (McKenzie et al, 2009). In respect of the latter, at equivalent absolute lung volumes the diaphragm pressure-generating capacity of patients with COPD is equal, or superior, to that of control participants (Similowski et al, 1991). However, despite this, the ability of the diaphragm to generate changes in volume at high lung volumes is diminished (McKenzie et al, 2009). It is important to keep in mind that diaphragm function at the same relative lung volumes is impaired in patients with COPD (see above), and that they have a reduced reserve capacity for volume and flow generation.
Change in diaphragm length is not the only chronic adaptation to hyperinflation and chronic inspiratory loading in patients with COPD. There are also changes in diaphragm biochemistry that appear to result from chronic loading (Levine et al, 1997; Ottenheijm et al, 2005). The healthy diaphragm is composed predominantly of two types of muscles fibres: one with high endurance but low power (type I, 45%), the other with low endurance but high power (type II, 55%). Patients with long-standing COPD have an abnormally high proportion of the former (type I 64%, type II 36%), which is an adaptive response to continuous inspiratory muscle loading (Levine et al, 1997).
Studies of the functional properties of the COPD-adapted diaphragm in vitro indicate that the fibres have a smaller cross-sectional area, contain less contractile protein and generate lower forces than those from patients without COPD (Ottenheijm et al, 2005). The dynamic properties of the contractile machinery of the COPD-adapted fibres are also impaired; the fibres appear to be less sensitive to calcium and show slower rates of myosin to actin attachment / detachment (Ottenheijm et al, 2005). Thus there is not only a loss of contractile protein; the protein that remains is also dysfunctional.
On the face of it, a shift towards an endurance-trained phenotype might be considered a positive adaptation; indeed it is cited as a reason for the futility of specific inspiratory muscle training (Polkey et al, 2011). However, it has been suggested that the increase in the proportion of type I fibres might, at least in part, explain the reduction in force-generating capacity (Clanton & Levine, 2009). Thus, depending upon the specific demands placed upon the inspiratory muscles, this adaptation can be either advantageous or disadvantageous. For example, it is advantageous for prolonged, low-intensity work, but disadvantageous for short, high-intensity work. The former is encountered at rest, whereas the latter is encountered during exercise. The diaphragm in patients with COPD therefore appears to be well adapted to generating low flow rates for long periods of time, but this adaptation robs them of the ability to generate the high pressures and flow rates required during exercise.
This suggestion is confirmed by studies of the in vivo strength and endurance of the inspiratory muscles of patients with COPD. For example, compared with control individuals, evoked diaphragm twitch pressure, maximal inspiratory pressure and a measure of endurance during inspiratory loading were all lower in patients with COPD (Barreiro et al, 2005). Furthermore, impairments were proportional to the severity of disease, despite the fact that a concomitant increase in type I fibres, and decrease in capillary to fibre ratio, were also proportional to disease severity. Thus, the shift towards a more endurance-trained phenotype reduced strength and did not appear to protect the inspiratory muscles from global fatigue under conditions of inspiratory loading (Barreiro et al, 2005). This is probably because weaker muscles must operate at a greater proportion of their maximum capacity, which predisposes them to fatigue.
Notwithstanding this apparent predisposition to fatigue, studies have so far failed to demonstrate evidence of exercise-induced contractile fatigue of the diaphragm in patients with COPD using low-frequency phrenic nerve stimulation (Polkey et al, 1995; Mador et al, 2000a; Mador et al, 2000b). However, this finding should not be misinterpreted to indicate that the inspiratory muscles are working within the limits of their capacity to deliver 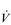 E, or that they do not impose any limitation upon exercise tolerance. The latter issue will be explored in greater detail in the section ‘Respiratory muscle involvement in exercise limitation’, but in the meantime it is noteworthy that studies where COPD patients walk (Kyroussis et al, 1996) or cycle (Yan et al, 1997) to the limit of tolerance have found a predominance of the rib cage muscle contribution to breathing. By measuring the rate of relaxation of the inspiratory muscles following a sniff effort, it is possible to detect the presence of global inspiratory muscle fatigue. Using this technique, it has been shown that, in patients who walk to the limit of tolerance, there is a slowing of the relaxation rate of oesophageal sniff pressure without any change in diaphragm twitch pressure, which is suggestive of accessory inspiratory muscle fatigue (Kyroussis et al, 1996). Furthermore, there does appear to be a subgroup of COPD patients who display diaphragm fatigue post-exercise (see below) (Hopkinson et al, 2010).
E, or that they do not impose any limitation upon exercise tolerance. The latter issue will be explored in greater detail in the section ‘Respiratory muscle involvement in exercise limitation’, but in the meantime it is noteworthy that studies where COPD patients walk (Kyroussis et al, 1996) or cycle (Yan et al, 1997) to the limit of tolerance have found a predominance of the rib cage muscle contribution to breathing. By measuring the rate of relaxation of the inspiratory muscles following a sniff effort, it is possible to detect the presence of global inspiratory muscle fatigue. Using this technique, it has been shown that, in patients who walk to the limit of tolerance, there is a slowing of the relaxation rate of oesophageal sniff pressure without any change in diaphragm twitch pressure, which is suggestive of accessory inspiratory muscle fatigue (Kyroussis et al, 1996). Furthermore, there does appear to be a subgroup of COPD patients who display diaphragm fatigue post-exercise (see below) (Hopkinson et al, 2010).
Finally, this section would be incomplete without mentioning the expiratory muscles, as well as contextualizing the changes in muscle function induced by COPD. As has already been alluded to, there is generalized muscle weakness, which also affects the expiratory muscles (Gosselink et al, 2000). In COPD patients the voluntary force-generating capacity of the expiratory muscles (maximal expiratory pressure: MEP) is ~ 30% lower than in healthy elderly people (Gosselink et al, 2000). This compares with differences in maximal inspiratory pressure (MIP), handgrip and quadriceps strength of ~ 40%, ~ 20% and 25%, respectively (Gosselink et al, 2000). The slightly larger effect of COPD upon MIP than MEP is most likely a manifestation of the additional influence of secondary weakness, due to the effects of hyperinflation (Gosselink et al, 2000). A recent study examined the influence of symptom limited cycling upon non-voluntary measures of expiratory and inspiratory muscle strength in patients with COPD; a significant exercise-induced fatigue of the abdominal muscles (7.2% fall in twitch gastric pressure) was found, but no change in diaphragm function (Hopkinson et al, 2010). Interestingly, only around one-third of the group exhibited expiratory muscle fatigue (twitch gastric pressure, 21%), and this subgroup also exhibited a significant fall in twitch diaphragm pressure (7.9%). The non-fatiguers exhibited no change in twitch gastric pressure, but a 7.7% increase in twitch diaphragm pressure. Unfortunately, the group was not subdivided to examine the diaphragm fatiguers in more detail. These data suggest that: (1) there is both inspiratory and expiratory muscle overload in at least some patients with COPD, and (2) diaphragm fatigue may be masked by lack of reliability in baseline measurements of twitch diaphragm pressure.
Patients with COPD also experience an increase in the demand for inspiratory muscle work, which arises from an elevated demand for minute ventilation (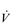 E), especially during exercise. Ventilation/perfusion mismatching and a higher than normal ratio of dead space to tidal volume (VD / VT) both necessitate an increase in
E), especially during exercise. Ventilation/perfusion mismatching and a higher than normal ratio of dead space to tidal volume (VD / VT) both necessitate an increase in 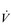 E in order to minimize changes in blood gases, but hypoxaemia is nevertheless a common finding. Furthermore, patients with COPD also have poor aerobic fitness, which increases the ventilatory demand of exercise (Casaburi et al, 1991), and thus increases inspiratory muscle work still further. Needless to say, these increased ventilatory flow requirements also exacerbate hyperinflation (Somfay et al, 2002).
E in order to minimize changes in blood gases, but hypoxaemia is nevertheless a common finding. Furthermore, patients with COPD also have poor aerobic fitness, which increases the ventilatory demand of exercise (Casaburi et al, 1991), and thus increases inspiratory muscle work still further. Needless to say, these increased ventilatory flow requirements also exacerbate hyperinflation (Somfay et al, 2002).
In summary, patients with COPD have a dramatically increased demand for inspiratory muscle work, but a reduced capacity to supply this demand due to muscle dysfunction. In other words, the demand / capacity relationship is stacked in completely the wrong direction. In the section ‘Respiratory muscle involvement in exercise limitation’, respiratory muscle-induced limitations to exercise tolerance will be considered, and Chapter 4 will review the evidence supporting specific respiratory muscle training for patients with COPD.
Asthma
The mechanical abnormalities in patients with asthma mimic closely those described in COPD; however, there are important differences. For example, there is less reduction in static lung recoil pressure and more widespread intrathoracic airway narrowing in asthma (Pride & Macklem, 1986). In addition, the increased airway collapsibility in patients with COPD is not seen in asthmatics. Furthermore, the reversible nature of airways obstruction in asthma results in relatively short-lived periods of stress upon the inspiratory muscles. The latter means that patients with asthma do not show the same changes in inspiratory muscle length or fibre composition that are expressed in patients with COPD (see above).
There is no clear consensus regarding the presence of primary weakness of the inspiratory muscles in patients with asthma compared with healthy people, as no biopsy data exists. However, the finding that steroid-dependent patients receiving oral corticosteroids show lower inspiratory muscle strength, but similar severity of hyperinflation, suggests that there may be myopathy in steroid-dependent patients with asthma (Akkoca et al, 1999). Generally, respiratory muscle strength and endurance are relatively normal in patients with stable asthma (Hill, 1991).
However, it is accepted universally that bronchoconstriction-induced hyperinflation is associated with secondary weakness of the inspiratory muscles (Fig. 3.1 inset) (Weiner et al, 1990; Perez et al, 1996; Akkoca et al, 1999; Stell et al, 2001; Weiner et al, 2002). As is the case in COPD, the major mechanical consequences of airway narrowing are increased flow resistive work, increased elastic work and PEEPi (resulting from dynamic lung hyperinflation), as well as reduced dynamic lung compliance (Martin et al, 1980; Lougheed et al, 1995). In a study comparing inspiratory muscle function of patients with COPD and asthma, with equivalent severity of hyperinflation, endurance was impaired to a greater degree in patients with asthma (Perez et al, 1996). Interestingly, strength was lower in the COPD patients compared with those with asthma. These data suggest that some of the structural and biochemical adaptations that occur in response to chronic loading in COPD are absent in patients with asthma. Thus, where airway obstruction is present, patients with asthma experience the same acute functional defect in their pulmonary function as those with COPD. However, the reversible nature of the airway obstruction may place patients with asthma at a functional disadvantage, and thus greater vulnerability to functional overload.
In a study of histamine-induced bronchoconstriction (FEV1 49% of baseline), the inspiratory work was found to increase 11-fold, 69% of the increase being due to the elastic component of the work of breathing (Martin et al, 1983). In addition, there also appears to be a prolonged activation of inspiratory muscles during exhalation in the presence of bronchoconstriction-induced hyperinflation (Muller et al, 1980; Muller et al, 1981), which suggests that the work of the total inspiratory muscles may be increased to an even greater extent than inspiratory work alone indicates.
The interrelationship between bronchoconstriction, hyperinflation and dyspnoea has also been studied. Multiple regression analysis indicates that, during methacholine-induced bronchoconstriction, change in inspiratory capacity (an index of dynamic hyperinflation) was the most powerful predictor of dyspnoea during bronchoconstriction – accounting for 74% of the variance in the perceptual rating (Lougheed et al, 1993). These observations are supported by more recent evidence confirming that hyperinflation is a major determinant of dyspnoea in patients with asthma (Martinez-Moragon et al, 2003).
As is the case in COPD, the mechanical changes associated with bronchoconstriction most likely increase the intensity of dyspnoea via their effect upon the magnitude of inspiratory neural drive (see Ch. 1). There is experimental support for this suggestion; Bellofiore et al (1996) found that the strongest determinant of dyspnoea during methacholine-induced bronchoconstriction was inspiratory neural drive (P0.1, mouth occlusion pressure), which explained 82% of the total variance in dyspnoea. More recently, Binks et al (2002) reported that institution of mechanical ventilation during methacholine-induced bronchoconstriction and hyperinflation significantly reduced ratings of ‘effort to breathe’ in people with mild asthma. Furthermore, it has also been shown that gender differences in inspiratory muscle strength may underpin differences in dyspnoea perception, quality of life and consumption of β2-agonist medication (Weiner et al, 2002). These data, along with data from inspiratory muscle training studies (see Ch. 4), support the notion that inspiratory muscle strength, and hence the relative intensity of inspiratory muscle work, makes a fundamental contribution to dyspnoea in people with asthma.
Because exercise is a trigger for asthma in around 90% of people with asthma (Wilkerson, 1998) there is an understandable anxiety regarding exercise that might translate into avoidance of physical activity, and poor aerobic fitness (Welsh et al, 2004). However, there remains no clear consensus regarding levels of physical activity and fitness, especially in children with asthma (Wilkerson, 1998), though there is some evidence to suggest that the aerobic fitness of adults with asthma is generally low (Satta, 2000). Thus, poor aerobic conditioning may exacerbate hyperinflation-related increases in the work of breathing during exercise by increasing the ventilatory requirement and exacerbating hyperinflation.
In summary, patients with asthma have an increased demand for inspiratory muscle work, which is proportional to the severity of their airway obstruction. It is not clear whether they have any primary weakness of their inspiratory muscles, but there is evidence of steroid-induced myopathy of the inspiratory muscles in steroid-dependent asthma. Furthermore, secondary weakness due to the influence of hyperinflation is well established, and linked strongly to dyspnoea. In the section ‘Respiratory muscle involvement in exercise limitation’, respiratory muscle-induced limitations to exercise tolerance will be considered, and Chapter 4 will review the evidence supporting specific respiratory muscle training for patients with asthma.
Bronchiectasis
Bronchiectasis is a chronic lung disease that is not normally included within the umbrella of COPD, but which overlaps with it (Neves et al, 2011); indeed one study found that 50% of patients with COPD also had bronchiectasis (Patel et al, 2004). It is characterized by irreversible widening of the medium-sized airways accompanied by inflammation, chronic infection and destruction of the bronchial walls (Neves et al, 2011). Both the pathology and the functional manifestations of bronchiectasis have similarities with those of COPD, including inflammatory cell profiles, protease release and consequent airway obstruction (Neves et al, 2011). In both conditions, these factors lead to detrimental changes in breathing mechanics, attendant exertional dyspnoea and exercise intolerance. Symptomology is also similar to COPD – including cough, sputum production and wheeze (Neves et al, 2011). Expiratory flow limitation (identified using the negative expiratory pressure technique) is present at rest in 39% of patients with bronchiectasis, which is a lower prevalence than in patients with COPD (Koulouris et al, 2003). The explanation for the latter finding may be that around half of patients had both obstructive and restrictive defects, i.e., restriction acted as a confounding influence; the presence of flow limitation was correlated with the MRC dyspnoea score, which in turn was correlated with exercise tolerance (Koulouris et al, 2003). Thus, the mechanical changes associated with bronchiectasis increase the demand for inspiratory muscle work, which is manifested symptomatically as exertional dyspnoea.
Compared with healthy people of a similar age, patients with moderate-to-severe bronchiectasis exhibit lower maximal inspiratory and expiratory muscle strength (around 20% and 40% lower, respectively) (Newall et al, 2005; Moran et al, 2010). The origin of this weakness is unclear, but is most likely due to a combination of primary weakness and functional weakness due to hyperinflation. Thus, in common with patients with COPD, patients with bronchiectasis have an imbalance in the demand / capacity relationship of the respiratory muscles. This imbalance will also be considered in the section ‘Respiratory muscle involvement in exercise limitation’, and Chapter 4 will review the evidence supporting specific respiratory muscle training for patients with bronchiectasis.
Cystic fibrosis
Respiratory failure is the most common cause of death in patients with cystic fibrosis (CF) (Taylor-Cousar, 2009), and dyspnoea is one of their main complaints (Leroy et al, 2011); it has also been suggested that the deterioration of lung function in patients with CF have is insufficient to explain their exertional dyspnoea. Patients with CF have an elevated work of breathing (Dunnink et al, 2009), and this has been identified as an important contributor to dyspnoea (Leroy et al, 2011). There appears to be no evidence of inspiratory muscle weakness in patients with CF; indeed, some authors have reported that patients with CF have superior strength (Dufresne et al, 2009; Dunnink et al, 2009) and diaphragm thickness (Dufresne et al, 2009). The elevated airway resistance of patients with CF appears to contribute to their diaphragm hypertrophy (Dufresne et al, 2009). However, patients with the lowest fat-free mass exhibit a loss of diaphragm thickness (Ionescu et al, 1998; Enright et al, 2007). Furthermore, although indices of inspiratory muscle strength have been found to be normal or superior in patients with CF, loss of maximal inspiratory muscle work capacity has been reported (Ionescu et al, 1998; Enright et al, 2007), suggesting that there is a deterioration in the metabolic properties of the inspiratory muscles. This finding is suggestive of an imbalance between demand and capacity since the preservation of inspiratory muscle strength is accompanied by an increased demand for inspiratory muscle work, and dyspnoea. The fact that respiratory failure is the primary cause of death highlights the important influence of the imbalance between demand and capacity. This will also be considered in the section ‘Respiratory muscle involvement in exercise limitation’, and Chapter 4 will review the evidence supporting specific respiratory muscle training for patients with CF.
Restrictive chest wall disorders
Conditions such as kyphoscoliosis, fibrothorax, thoracoplasty, flail chest and ankylosing spondylitis all induce chest wall restriction, creating a restrictive pulmonary defect in which total respiratory system elastance and resistance are elevated (Donath & Miller, 2009). In the case of severe kyphosis and / or scoliosis, thoracic volume may also be reduced by collapse of the vertebral column and the cranial displacement of the abdominal contents. As a consequence, breathing pattern tends to be rapid and shallow, creating a higher than normal ratio of dead space to tidal volume (VD / VT) ratio and necessitating an increase in 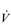 E. This exacerbates the already elevated work of inhalation (Donath & Miller, 2009), and attendant dyspnoea. Furthermore, inspiratory muscle function also tends to be impaired (Lisboa et al, 1985; Cejudo et al, 2009), owing to changes in chest wall and diaphragm configuration. In kyphoscoliosis, inspiratory muscle strength has been shown to correlate with forced vital capacity (FVC), as well as to arterial blood gases, such that weakest patients exhibited the worst FVC and blood gases (Lisboa et al, 1985). Ultimately, the outcome of these conditions can be respiratory failure and the requirement for mechanical ventilation. The imbalance in the demand / capacity relationship of the respiratory muscles will also be considered in the section ‘Respiratory muscle involvement in exercise limitation’. See Chapter 4 for a description of the evidence supporting breathing exercises.
E. This exacerbates the already elevated work of inhalation (Donath & Miller, 2009), and attendant dyspnoea. Furthermore, inspiratory muscle function also tends to be impaired (Lisboa et al, 1985; Cejudo et al, 2009), owing to changes in chest wall and diaphragm configuration. In kyphoscoliosis, inspiratory muscle strength has been shown to correlate with forced vital capacity (FVC), as well as to arterial blood gases, such that weakest patients exhibited the worst FVC and blood gases (Lisboa et al, 1985). Ultimately, the outcome of these conditions can be respiratory failure and the requirement for mechanical ventilation. The imbalance in the demand / capacity relationship of the respiratory muscles will also be considered in the section ‘Respiratory muscle involvement in exercise limitation’. See Chapter 4 for a description of the evidence supporting breathing exercises.
Interstitial lung disease (ILD) is an umbrella term for a group of lung disorders that share a number of pathophysiological characteristics and clinical features. The principal feature of ILD is exercise intolerance due to exertional dyspnoea and perceptions of fatigue. Exercise intolerance is correlated with quality of life (Holland, 2010), which makes it an important therapeutic target.
The reduced lung compliance in ILD leads to impairment of vital capacity, and a rapid and shallow breathing pattern that worsens during exercise (Javaheri & Sicilian, 1992). This pattern exacerbates the existing ventilation / perfusion (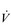 /
/ 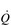 ) mismatch, due to its effect upon the VD / VT ratio. There is also an impairment of diffusing capacity, and the combination with
) mismatch, due to its effect upon the VD / VT ratio. There is also an impairment of diffusing capacity, and the combination with 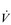 /
/ 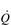 mismatching can precipitate substantial arterial desaturation (Miki et al, 2003). These changes also increase the ventilatory demand of exercise and hence the work of breathing.
mismatching can precipitate substantial arterial desaturation (Miki et al, 2003). These changes also increase the ventilatory demand of exercise and hence the work of breathing.
Sarcoidosis involves multiple organs, but pulmonary manifestations typically predominate (Lynch et al, 2007) in the form of an ILD. Dyspnoea is the most common presentation in patients with early to moderately advanced disease (Baydur et al, 2001). Sarcoidosis is associated with reduced inspiratory and expiratory muscle strength (~ 20% reduction), as well as impaired endurance (Wirnsberger et al, 1997; Baydur et al, 2001; Spruit et al, 2005), and respiratory muscle function correlates more closely with dyspnoea during activities of daily living than pulmonary function (Baydur et al, 2001); indeed dyspnoea can be present in the absence of any lung function defects (Baydur et al, 2001). The underlying mechanisms for respiratory muscle dysfunction in sarcoidosis are unclear, but two case reports indicate that granulomatous involvement of respiratory muscles is present (Dewberry et al, 1993; Pringle & Dewar, 1997). At the time of writing there have been no studies of respiratory muscle training in patients with sarcoidosis or other ILD.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


