Chapter 3 The Pathogenesis of SLE
The Phases of Sle: Evolution of Disease in Susceptible Persons
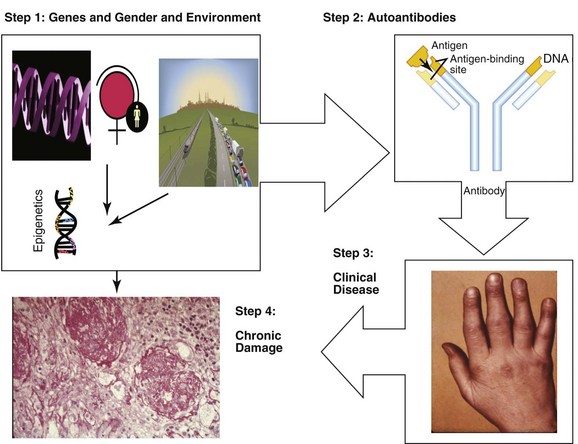
As shown in Figure 3-1, the development of SLE occurs in a series of steps. There is a long period of predisposition to autoimmunity, conferred by genetic susceptibility, gender, and environmental exposures, and then (in a small proportion of those predisposed) development of autoantibodies, which usually precede clinical symptoms by months to years. A proportion of individuals with autoantibodies demonstrate clinical SLE, often starting with involvement of a small number of organ systems or abnormal laboratory values, and then evolving into enough clinical and laboratory abnormalities to be classified as SLE. Finally, over a period of many years, most individuals with clinical SLE experience intermittent disease flares and improvements (usually not complete remission), and compile organ damage and comorbidities related to genetic predisposition, chronic inflammation, activation of pathways that damage organs (such as renal tubules), and/or induce fibrosis, to therapies, and to aging.
Overview: The Major Immune Pathways Favoring Autoantibody Production
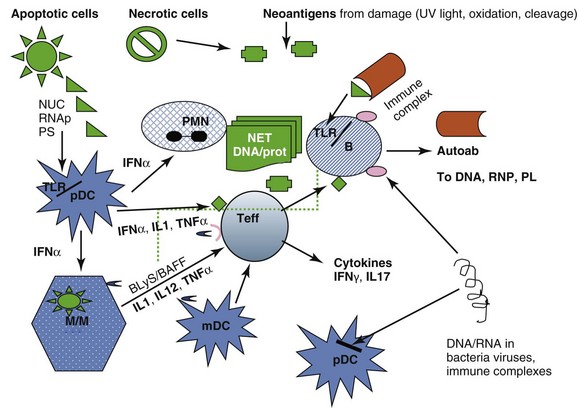
These pathways are summarized in Figure 3-2.
Stimulation of Innate and Adaptive Immune Responses by Autoantigens
The autoantigen stimulation of the innate and adaptive immune responses is provided by cells undergoing apoptosis (which present autoantigens such as nucleosome and Ro in surface blebs, and phosphatidyl serine on outer surfaces of membranes), by cells undergoing necrosis and releasing cell components which can form neoantigens under the influence of oxidation, phosphorylation, and cleavage, and by microorganisms that have antigenic sequences that cross-react with human autoantigens. Antigen-presenting cells—dendritic cells (DCs), monocytes/macrophages (M/Ms), and B lymphocytes process and present such antigens (Ags). In addition, cells of innate immunity (DC, M/M) are activated via internal Toll-like receptors (TLRs) by DNA/protein and RNA/protein that can be provided by dying cells, particularly polymorphonuclear neutrophils (PMNs) undergoing NETosis, by SLE immune complexes (ICs), and by infectious agents. The net result of activation of DCs from tolerogenic to proinflammatory cells secreting inflammatory cytokines (including the lupus-promoting interferon alpha [IFN-α]), and of M/Ms to proinflammatory cells secreting tumor necrosis factor alpha (TNF-α), and interleukins IL-1, IL-12, and IL-23, is activation of effector T cells that help B cells make immunoglobulin (Ig) G autoantibodies, infiltrate tissues, and be cytotoxic for some tissue cells such as podocytes in the kidney. B lymphocytes, activated directly by DNA/protein and RNA/protein via their TLRs and by IFN-α, can also be helped in their secretion of autoantibodies by T cells, and in their survival and maturation to plasmablasts by BLyS (B-lymphocyte stimulator)/BAFF (B cell–activating factor), IL-6, and other cytokines. In patients with SLE these processes escape normal regulatory mechanisms, which are listed in Box 3-1. Thus, autoantibodies induce the first phase of clinical disease (organ inflammation of joints, skin, glomeruli, destruction of platelets, etc.) because (1) the autoantibodies and the ICs they form persist, (2) they are quantitatively high, (3) they contain subsets that bind target tissues, (4) they form immune complexes that are trapped in basement membranes or bound on cell surfaces, (5) charges on antibodies or ICs favor nonspecific binding to tissues, and (6) their complexes activate complement. And yet, in spite of this deluge of autoantibodies and ICs attacking tissue, mouse models suggest that susceptibility to clinical disease requires more—there are several examples of autoantibody formation, abundant Ig deposition in glomeruli, and complement fixation without development of clinical nephritis.
Box 3-1
Mechanisms of Downregulation of the Immune Response That Are Defective in SLE
1. Disposal of immune complexes (ICs) and apoptotic cells (ACs): Defective phagocytosis, transport by complement receptors, and binding by Fcγ receptors. Can be due to macrophage defects intrinsic to SLE, low levels of complement-binding CR1 receptors—or occupied receptors, FcγRs that are occupied, downregulated, or genetically low-binding of the immunoglobulin (Ig) in ICs. Early components of complement or mannose-binding lectin/ protein (MBL) also participate in solubilizing and transporting IC. They may be missing or defective.
2. Defective idiotypic networks: due to low production of anti-idiotypic antibodies, defective regulation of T helper cells by T-regulator cells that recognize idiotypes in their T-cell receptors (TCRs).
3. Inadequate production and/or function of regulatory cells that kill or suppress autoreactive B cells, T helper cells, other effector cells. This includes CD8+ cytotoxic cells that kill autoreactive B, regulatory CD4+CD25+Foxp3+ T cells that normally target both T helper cells and autoreactive B cells, inhibitory CD8+Foxp3+ T cells that suppress both T helper and B cells, regulatory B cells, and tolerogenic dendritic cells (DCs). Possibly natural killer (NK) cell defects.
4. Low production of interleukin-2 (IL-2) by T cells. Survival of regulatory T cells requires IL-2, and effector T cells in SLE make decreased quantities of IL-2. IL-2 is also required for activation-induced death in lymphocytes.
5. Defects in apoptosis that permit survival of effector T and autoreactive B cells, usually genetically determined.
Regulatory Mechanisms Fail to Control Autoimmune Responses
As shown in Box 3-1, several mechanisms that downregulate active immune responses are defective in SLE.
Abnormalities in T and B Lymphocytes in SLE
T cells in SLE are also abnormal. Like B cells, they respond to lesser stimuli than are required for healthy T cells. A major abnormality of SLE CD4+ T cells is assembly of an abnormal signaling apparatus after T-cell receptor (TCR) activation. Figure 3-3 shows some of these abnormalities. In health, TCR stimulation results in assembly of the CD3ζ chain into the surface activation cluster. In SLE, the FcRγ chain is substituted for CD3ζ, resulting in a different activation pathway. The end results are increased release of intracellular calcium, which promotes translocation of calcium/calmodulin-dependent protein kinase IV (CaMK4) to the nucleus, and upregulation of transcription repressor cyclic adenosine monophosphate (AMP) response–element modulator alpha (CREM-α), which on binding to promoter regions of DNA suppresses IL-2 production and enhances IL-17 production. Abnormally low secretion of IL-2 by T cells impairs production of regulatory T cells, whereas increased production of IL-17 promotes inflammation. Causes of the downregulation of CD3ζ include antibodies to T lymphocytes and mTOR activation in T cells resulting from increased levels of nitric oxide (NO) and elevated transmembrane potentials in the mitochondria of SLE T cells. SLE T-cell subsets have many other abnormalities: CD8+ cytotoxic T cells may be defective, adding to the persistence of autoreactive B cells. Regulatory T cells of CD4+ and CD8+ phenotypes also are abnormal in quantities and/or functions. Double-negative (DN) T cells (CD3+CD4−CD8−), which probably derive from CD8+ T cells, infiltrate tissue and secrete IL-17.
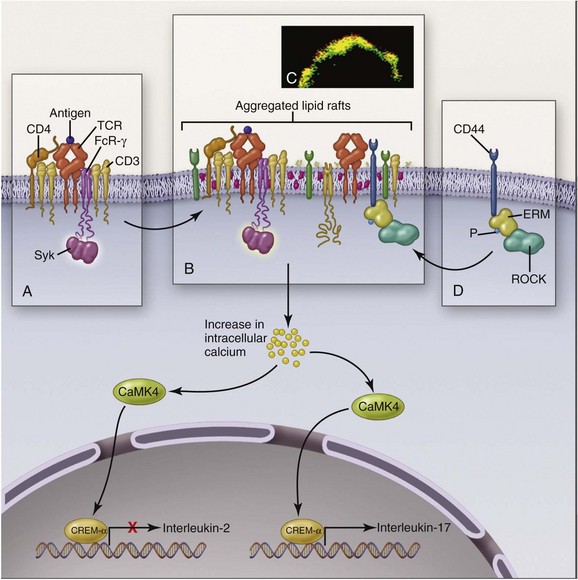
FIGURE 3-3 Abnormalities of T lymphocyte activation in patients with SLE.
(From Tsokas GC: Systemic lupus erythematosus. N Eng J Med 365:2110–2121, 2011.)
Related posts:
 The Innate Immune System in SLE
Clinical Aspects of the Antiphospholipid Syndrome
Gastrointestinal and Hepatic Manifestations
Novel Therapies for SLE: Biological Agents Available in Practice Today
Mixed Connective Tissue Disease and Undifferentiated Connective Tissue Disease
Pathomechanisms of Cutaneous Lupus Erythematosus
The Innate Immune System in SLE
Clinical Aspects of the Antiphospholipid Syndrome
Gastrointestinal and Hepatic Manifestations
Novel Therapies for SLE: Biological Agents Available in Practice Today
Mixed Connective Tissue Disease and Undifferentiated Connective Tissue Disease
Pathomechanisms of Cutaneous Lupus Erythematosus
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


