Chapter 20 Autoantibodies
Antibody Structure and Function
All antibody molecules have a common core structure of two identical light chains and two identical heavy chains. The four chains are connected by disulfide bonds—each heavy chain to a light chain, and the heavy chains to each other. These four chains fold to form a globular motif, hence the term immunoglobulin (Figure 20-1).
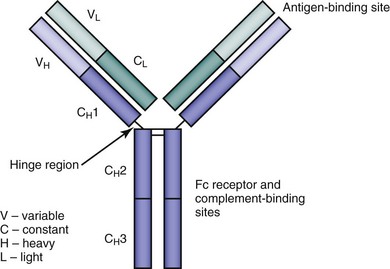
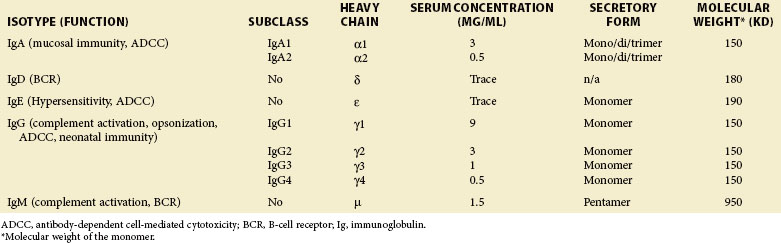
Human antibody molecules are divided into five classes, or isotypes, IgA, IgD, IgE, IgG, and IgM, on the basis of their heavy chains. IgA and IgG molecules are further divided into subclasses. It is the heavy chain isotype of each antibody molecule that determines its effector mechanisms. There are two light chain isotypes, κ and λ, although no differences in function have been identified. In humans, usage of κ and λ light chains is approximately equal, but in mice the ratio is more like 10:1.1 The structure and function of the different antibody isotypes are described in Table 20-1.
Early investigation of immunoglobulin structure utilized the proteolytic enzyme papain.2 Treatment with papain leads to disruption of the hinge region of the immunoglobulin molecule (see Figure 20-1) and the production of three fragments. Two of the fragments are identical, each consisting of a light chain plus the variable and first constant regions of a heavy chain, and were thus termed the antigen-binding fragments (Fab). The remaining fragment was noted to crystallize easily into diamond-shaped plates, and accordingly was called the Fc fragment.
Antibody Production and the Generation of Diversity
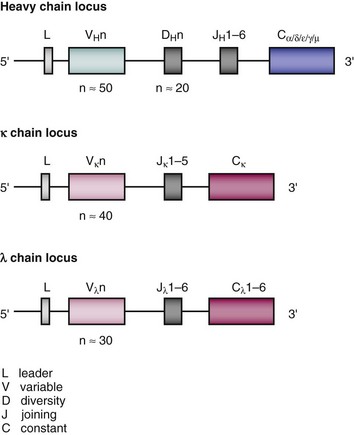
Loci for the different immunoglobulin chains are found on separate chromosomes.1 In humans, the κ light (L) chain locus is on chromosome 2, the λ light chain locus on chromosome 22, and the heavy (H) chain locus on chromosome 14. Each locus contains multiple genes encoding the variable (V) and constant (C) regions. At the 5′ end of the locus are the V genes, and in humans, each heavy and light chain locus contains 100- to 200-V genes. However, many of these are nonfunctioning pseudogenes, and the actual number of genes that can be used is more like 50 VH, 40 Vκ, and 30 Vλ. There is 1 gene in the human κ C region locus, and 3 to 6 genes in the λ locus. The heavy chain locus contains all the genes necessary to produce the different immunoglobulin isotypes. Between the V and C regions, there are additional genes named the joining (J) and diversity (D) segments. The latter are found only in the heavy chain locus. The third CDR of the variable chain is encoded by genes from the V region and the J/D segments. The structure of the human immunoglobulin chain loci are shown in Figure 20-2.
The enormous diversity in antibody specificity is achieved by a number of mechanisms. As discussed, there are multiple possible V, D, and J germline genes, and variability is further increased by different VDJ and different heavy and light chain combinations. In addition, small changes in the nucleotide sequences of junctional regions (i.e., between V, D, and J genes) occur from either imprecisions in the rearrangement mechanism or the addition of new nucleotides. Finally, after antigen is encountered, further diversity is achieved by somatic mutation, which accounts for affinity maturation of antibody, as described previously.1,3
1 Abbas AK, Lichtman AH, Pober JS. Cellular and molecular immunology, ed 2. Philadelphia: WB Saunders; 1994.
2 Porter RR. Structural studies of immunoglobulins. Science. 1973;180:713–716.
3 Janeway CA, Travers P, Walport M, et al. Immunobiology: the immune system in health and disease, ed 5. New York: Garland Science; 2001.
Antibodies to DNA, Histones, and Nucleosomes
Anti-DNA Antibodies in Lupus: Historical Overview
Anti-DNA antibodies were the first autoantibodies described in patients with systemic lupus erythematosus (SLE), having been reported by four separate research groups in 1957 (reviewed in reference 1). Over the next 50 years, evidence obtained through a variety of approaches suggested that these antibodies were important in the pathogenesis of the disease, that is, that they had direct and damaging effects on tissues. This suggestion is particularly true of antibodies to double-stranded DNA (anti-dsDNA) as opposed to single stranded DNA (anti-ssDNA).
Some of this evidence was derived from serologic studies in cohorts of patients with SLE. These studies showed that whereas anti-dsDNA antibodies can be found in up to 70% to 80% of patients with SLE at some time during the course of their disease, these antibodies are very rarely found in patients with other autoimmune conditions and in healthy controls. Using stored samples taken from American military recruits in whom SLE later developed, Arbuckle showed that in some cases anti-dsDNA antibodies are present several years before the onset of clinical disease.2
Furthermore, in many cases there is a relationship between disease activity and the serum titer of anti-dsDNA. For example, in an early study Schur and Sandson looked at serum samples taken from 96 patients with SLE (44 with nephritis) over a 2-year period.2a Anti-dsDNA and anti-ssDNA were found in more than 60% of patients with active nephritis but in only 10% to 15% of those with inactive disease. Characteristically, exacerbations of nephritis in these patients were preceded by the appearance of anti-DNA antibodies and a drop in serum complement. In a larger study, Swaak showed that a rise in levels of anti-dsDNA antibodies preceded renal flares in SLE,3 and subsequent independent studies also showed that rises in anti-dsDNA antibody level were associated with flares of activity, either in the kidney or in other organs. In fact, one of the main disease activity indices in SLE—the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) actually includes raised anti-dsDNA antibodies as a scorable element of disease activity. Both isotype and binding properties of anti-DNA antibodies affect association with disease activity. Immunoglobulin (Ig) G anti-dsDNA antibodies are particularly important. Perhaps the clearest evidence for their importance came from a Japanese study in which renal biopsies were carried out in 40 patients with untreated lupus nephritis and the histologic degree of nephritis was compared with levels of IgG and IgM antibodies to dsDNA and ssDNA.4 Levels of IgG anti-dsDNA were more closely correlated with nephritis than IgM anti-dsDNA or anti-ssDNA of either isotype.
In parallel with these serologic studies, other investigators showed that anti-DNA antibodies are present in inflamed organs of patients with SLE, with the clearest evidence relating to lupus nephritis. In a seminal experiment in 1967, Koffler showed deposition of IgG and complement in glomeruli of patients who had died from lupus nephritis.5 Eluates from these kidneys bound nuclei, and this binding could be partially inhibited by the addition of dsDNA.5 Winfield showed that glomerular eluates from autopsy specimens of patients who had died from lupus nephritis contained higher-avidity anti-dsDNA antibodies than serum from the same patients.6 It is important to note, however, that other autopsy studies have shown that antibodies with different specificities (e.g., Ro, La, Sm, and C1q) can also be eluted from glomeruli of patients with lupus nephritis.
Measurement of Anti-dsDNA Antibodies
New forms of ELISA in which the source of dsDNA and the reaction conditions have been optimized are reported to give specificity for high-avidity antibodies as good as that of the Farr assay and are likely to come into more widespread use in the future. In a longitudinal study of 16 patients with newly diagnosed lupus nephritis, Manson found that one of these assays showed the same degree of association with measures of renal disease (such as urine protein/creatinine ratio) as measurement of antinucleosome level.7 In fact there is no strong evidence that replacing the currently available anti-dsDNA ELISAs with antinucleosome assays would be beneficial in the management of the majority of patients with SLE. There is a subgroup of patients with persistently high anti-dsDNA despite having no disease activity (serologically active, clinically quiescent [SACQ]). In these patients with SACQ disease, Ng reported that high antinucleosome levels were associated with a higher number of disease flares and reduced time to first flare over the next 5 years.8 Thus, measuring antinucleosome levels might be worthwhile in patients with SACQ disease.
Work from Experimental Models Emphasing the Potential Importance of Anti-dsDNA Antibodies
Although it is important to study patients to ensure clinical relevance, strong evidence for a directly pathogenic role of anti-dsDNA antibodies comes from work using animal models. The major studies have been reviewed in detail elsewhere.1 Various groups have observed the effect of monoclonal anti-dsDNA antibodies in nonautoimmune mice strains. Ravirajan generated a human panel of anti-dsDNA antibody–producing hybridoma cells from the lymphocytes of patients with SLE.9 Following intraperitoneal implantation of hybridoma cells producing one such antibody, RH14, severe combined immunodeficient (SCID) mice went on to demonstrate significant proteinuria with human immunoglobulin deposition within the kidney. Furthermore, electron microscopy findings were very suggestive of lupus nephritis–like disease, with mesangial cell hypertrophy, mesangial and endothelial cell deposits, and podocyte foot process effacement. It is important to note, however, that these pathogenic effects are not always observed. A second DNA-binding antibody, generated from a different patient, and noted to have much less diverse antigen binding in vitro, produced only minimal proteinuria and did not deposit in the mouse kidney or engender the pathologic changes observed with RH14.
Raz, using an isolation rat kidney perfusion system, showed that some murine monoclonal anti-dsDNA antibodies (and affinity-purified human anti-dsDNA antibodies) were able to increase proteinuria significantly.10
There is a significant body of work that examines the effect that small changes in the antigen-binding site have on antibody-binding properties in vitro and in vivo. By studying panels of murine11 and human12 monoclonal anti-dsDNA antibodies, it has been shown that there is a high prevalence of arginine, asparagine, and lysine residues in the complementarity-determining regions (CDRs) of anti-dsDNA antibodies. It is proposed that the presence and position of these amino acids facilitate the antibody-DNA interaction. The accumulation of these particular amino acids is driven by somatic hypermutation—the accumulation of beneficial mutations, which increase antigen affinity and promote survival of the B-cell clone.
How Pathogenic Anti-dsDNA Antibodies Bind to Tissues: The Importance of Binding to Nucleosomes
The previous section summarized evidence that circulating anti-dsDNA antibodies are deposited in tissues such as the kidney and cause inflammation. The mechanism whereby this process occurs has been studied intensively, and there are a number of theories that are not mutually exclusive. Within a single patient, these antibodies may be deposited by different mechanisms in different tissues or even within a single tissue. Though initial theories proposed that immune complexes of anti-dsDNA with dsDNA would be deposited in tissues, this now seems unlikely because there is very little circulating free dsDNA in human serum. Analysis of the molecular weight of DNA found in the circulation of patients with SLE showed that it occurred in fragments of 200 bp (or multiples thereof). The explanation is that this dsDNA is in the form of oligonucleosomes formed as debris from the breakdown of apoptotic cells. Nucleosomes are the base units of nuclear chromatin and consist of approximately 200 bp of dsDNA wrapped round a histone core. The identification of nucleosomes,13 rather than free dsDNA, as the key antigen recognized by “anti-dsDNA” antibodies in SLE resolves a number of questions, as follows.
First, because dsDNA is present inside the nuclei of cells, it had been difficult to understand how anti-dsDNA antibodies could access their antigen. However, apoptotic cells release blebs in which previously intracellular antigens, such as nucleosomes, are exposed on the surface. The removal of this apoptotic debris is known to be slower in patients with SLE than in healthy controls,14 so that such patients possess larger amounts of circulating nucleosome material, which could interact with antinucleosome antibodies.
Second, the concept that the pathogenic antibodies in SLE circulate in the form of nucleosome/antinucleosome complexes provided a possible mechanism for deposition of these complexes in the kidney and skin. This mechanism has been described and investigated by a Dutch group, who propose that positively charged histones in the nucleosomes of the nucleosome/antinucleosome complex interact with negatively charged heparan sulfate in the renal basement membrane.15 In a series of elegant experiments in a perfused rat kidney model these investigators showed that deposition of IgG could be achieved by adding histones, then DNA, then antinucleosome antibody or by adding nucleosome/antinucleosome complexes generated in vitro. Deposition was reduced by prior perfusion with heparatinase, which removed heparan sulfate. The group has also shown deposition of antinucleosome antibodies in the skin of patients with lupus nephritis. Kalaaji has used electron microscopy to show that IgG in the kidneys of both patients with lupus nephritis and murine models of lupus colocalize with electron-dense extracellular deposits of chromatin, a finding consistent with the model that antibody-nucleosome interactions are important in the pathogenesis of lupus nephritis.16
Supporting these histologic findings are serologic studies showing that antinucleosome antibodies are present at a high prevalence in patients with SLE and may be related to disease activity. Studies by various groups have shown raised levels of antinucleosome antibodies in between 56% and 86% of patients with SLE.17 The specificity of this test depends on the purity of the nucleosomes used.
Cross-Reaction of Anti-DNA Antibodies with Intracellular Antigens
The antigen α-actinin-4 is produced by renal podocytes in glomeruli and plays an important role in the function of these cells. Point mutations in the α-actinin-4 gene cause a form of focal segmental glomerulosclerosis with nephrotic syndrome—although unlike lupus nephritis, this glomerulosclerosis is not characterized by deposition of antibodies and complement. Two groups working independently in mouse models of lupus showed that the ability of some murine monoclonal ANAs to cause glomerulonephritis after passive transfer depended on the ability to bind α-actinin rather than dsDNA. Subsequent clinical studies showed that anti–α-actinin antibodies are present in patients with SLE (though not specific for that disease) and that in some cohorts positivity for these antibodies might distinguish patients with and without renal involvement. However, most patients with lupus nephritis do not have anti–α-actinin antibodies, and a longitudinal study in 16 patients with newly diagnosed lupus nephritis showed that levels of anti-dsDNA and antinucleosome antibodies were much more closely associated with the presence of lupus nephritis and with clinical markers of renal function during the follow-up period than were levels of anti–α-actinin.7 Currently, therefore, interaction with nucleosomes seems likely to be the predominant mechanism for renal deposition of pathogenic antibodies in lupus nephritis.
Antihistone Antibodies
Though the antinucleosome antibodies just described can bind to histones, the term antihistone antibodies is generally used to refer to a different type of antibody detected by ELISA using histones as the test antigen. Antihistone positivity is particularly characteristic of patients with drug-induced lupus caused by exposure to drugs such as hydralazine and procainamide. In general these patients have mild disease with a low frequency of nephritis that remits when the drug is stopped. However, patients with spontaneous (non–drug-induced) SLE can also test positive for antihistone antibodies, as shown by Gioud in 32 of 63 patients with SLE but only 1 of 70 patients with other rheumatic diseases.18
Structure and Origin of Pathogenic Anti-dsDNA and Antinucleosome Antibodies
The fact that serologic studies showed that IgG anti-dsDNA antibodies were particularly closely related to disease activity in patients with SLE was supported by studies in which passive transfer of monoclonal human or murine anti-dsDNA antibodies caused glomerulonephritis in mice. In these studies, a consistent finding was that only some antibodies would cause nephritis. As previously described, in experiments in which six different hybridomas secreting monoclonal human IgG anti-dsDNA antibodies were introduced into severe combined immunodeficiency (SCID) mice, only two caused deposition similar to that seen in lupus nephritis.9 A number of groups carried out sequence analysis of monoclonal human12 and murine11 anti-DNA antibodies, some of which were pathogenic in mouse models. The conclusion was that pathogenicity was more likely to be a property of antibodies that had gone through the processes of class switching and somatic mutation. The somatic mutations in these antibodies were clustered in the CDRs, implying that they had been accumulated nonrandomly as a result of antigen drive. This process typically occurs in germinal centers.
A B-lymphocyte clone, in the presence of T-helper cells, is stimulated to divide by antigen interacting with its surface immunoglobulin. The greater the affinity of the surface antibody for antigen, the more powerful the stimulus to divide and the faster the clone grows. As the cells divide, some incorporate somatic mutations owing to a specific hypermutation mechanism that operates only in B lymphocytes at this stage of their development and acts only upon the rearranged immunoglobulin sequences. Thus the different B cells in the clone contain a range of expressed antibody sequences that differ only at sites of somatic mutation. Any B cell that picks up a mutation that enhances binding to the driving antigen is stimulated to divide more strongly than its neighbors and has more descendants. Thus over time, the clone becomes dominated by cells containing mutations at positions in the sequence that improve binding to antigen. These positions are usually in or around the CDRs, because the CDRs encode the antigen-binding site. The accumulation of these mutations leads to a gradual increase in the antigen-binding affinity of the antibody secreted by the clone (affinity maturation). Within anti-DNA antibodies, it appears that mutations to the residues arginine, asparagine, and lysine within CDRs are particularly important to facilitate binding to dsDNA,11,12 although this is not a universal rule and there are several examples of antibodies in which increased numbers of such residues are not associated with greater binding to dsDNA.
Which antigen drives the production of these high-affinity, somatically mutated IgG antibodies? The ideal antigen for this purpose would be one that is present in larger quantities in patients with SLE and that carries epitopes for both B cells and T-helper cells. Nucleosomes fit the bill. As noted previously, they are present on the surface of apoptotic blebs, which are not cleared quickly in patients with SLE.14 This material can therefore be carried to lymphoid tissues, where it can stimulate both T-helper cells (via histone epitopes) and B cells that can then secrete antinucleosome antibodies.
If the nucleosome or apoptotic material really do provide the antigenic stimulus for development of somatically mutated autoantibodies in SLE, one would expect reversal of the mutations to reduce affinity for those antigens. Evidence supporting this expectation was produced by experiments in which antibodies were expressed from cloned cDNA in vitro and the cloned DNA was then altered to allow expression of antibodies in which one or more somatic mutations had been reverted to the germline sequence. The properties of the expressed antibodies containing different numbers of somatic mutations were then compared, and several researchers reported that mutations at single sites could alter binding to apoptotic cells, dsDNA, and other antigens typically found on apoptotic blebs. Thus, Cocca, who reverted a single mutation from arginine to serine at position 53 in CDR3 of the heavy chain of the pathogenic murine anti-dsDNA antibody 3H9, found that this change simultaneously reduced strength of binding to DNA, phospholipids, beta 2-glycoprotein I, and apoptotic cells.19 Similarly, Wellman showed that three somatic mutations in CDRs of the pathogenic human anti-dsDNA antibody 33C9 were critical for binding to dsDNA, nucleosomes, and apoptotic cells.20
Can Measuring Anti-dsDNA Levels Help Us Manage Patients with SLE?
If levels of anti-dsDNA antibodies rise during periods of high disease activity, could we improve our management of patients with SLE by treating patients when these levels rise but before a clinical flare becomes apparent? The first trial to investigate this possibility was carried out by the Bootsma group in the mid-1990s.21 They followed a cohort of 156 patients with SLE and identified 46 who experienced a rise in dsDNA. These 46 patients were randomly allocated to receive either conventional treatment (i.e., the dose of steroids was increased only if clinical symptoms of a flare developed) or increased prednisolone dose right away (the increase was 30 mg/day, followed by tapering over 18 weeks). The group in whom prednisolone dose was increased straightaway whenever anti-dsDNA level rose did have significantly fewer flares of disease but also experienced more adverse effects of steroids, and more than one quarter of this group discontinued the trial. A later study using a shorter course of higher steroid dose suggested that patients in whom both anti-dsDNA and C3a levels rose experienced significantly fewer flares when treated with prednisolone than when treated with placebo.22
Could we treat patients with SLE by targeting the production of anti-dsDNA antibodies? Because antibodies are produced by B lymphocytes, anti–B cell therapies would be expected to reduce anti-dsDNA levels. Both the anti-CD20 agent rituximab23 and the anti–B lymphocyte stimulator agent belimumab24 have been studied in randomized placebo-controlled clinical trials. The belimumab trial did show a benefit of drug over placebo, whereas the rituximab trial did not, although there is still much discussion about possible reasons for this difference, including the different trial designs and outcome measures used in the two trials. A number of previous open studies had suggested that treatment with rituximab is effective in many patients with SLE and leads to a fall in anti-dsDNA levels.25 We await conclusive information about whether levels of anti-dsDNA antibodies can be used to guide selection of patients for treatment with either rituximab or belimumab.
In contrast, an ambitious attempt to treat SLE by specific deletion of B cells producing anti-dsDNA antibodies appears to have ended in failure. The therapeutic agent, called abetimus sodium, consisted of four oligonucleotide chains bound to an inert carrier. The theory was that the oligonucleotides would engage anti-dsDNA antibodies on the surface of B cells (and would thus not interact with any cells not producing anti-dsDNA antibodies). The lack of a T-cell epitope on the drug would mean that the engaged B cells could not recruit T-cell help and so would die. Treatment with abetimus sodium led to reduction of anti-dsDNA antibody levels and clinical improvements in a mouse model of SLE. In human trials, anti-dsDNA levels fell,26 but it was not possible to demonstrate any significant difference between drug and placebo in the ability to delay the onset of renal flare.
1 Isenberg DA, Manson JJ, Ehrenstein MR, et al. Fifty years of anti-ds DNA antibodies: are we approaching journey’s end? Rheumatology (Oxford). 2007;46:1052–1056.
2 Arbuckle MR, McClain MT, Rubertone MV, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349:1526–1533.
2a Schur PH, Sandson J. Immunologic factors and clinical activity in systemic lupus erythematosus. N Engl J Med. 1968;278:533–538.
3 Swaak AJ, Aarden LA, Statius van Eps LW, et al. Anti-dsDNA and complement profiles as prognostic guides in systemic lupus erythematosus. Arthritis Rheum. 1979;22:226–235.
4 Okamura M, Kanayama Y, Amastu K, et al. Significance of enzyme linked immunosorbent assay (ELISA) for antibodies to double stranded and single stranded DNA in patients with lupus nephritis: correlation with severity of renal histology. Ann Rheum Dis. 1993;52:14–20.
5 Koffler D, Schur PH, Kunkel HG. Immunological studies concerning the nephritis of systemic lupus erythematosus. J Exp Med. 1967;126:607–624.
6 Winifred JB, Faiferman I, Hoffler D. Avidity of anti-DNA antibodies in several IgG glomerular eluates from patients with systemic lupus erythematosus. Association of high avidity anti-native DNA antibody with glomerulonephritis. J Clin Invest. 1977;59:90–96.
7 Manson JJ, Ma A, Rogers P, et al. Relationship between anti-dsDNA, anti-nucleosome and anti-alpha-actinin antibodies and markers of renal disease in patients with lupus nephritis: a prospective longitudinal study. Arthritis Res Ther. 2009;11:R154.
8 Ng KP, Manson JJ, Rahman A, et al. Association of antinucleosome antibodies with disease flare in serologically active clinically quiescent patients with systemic lupus erythematosus. Arthritis Rheum. 2006;55:900–904.
9 Ravirajan CT, Rahman MA, Papadaki L, et al. Genetic, structural and functional properties of an IgG DNA-binding monoclonal antibody from a lupus patient with nephritis. Eur J Immunol. 1998;28:339–350.
10 Raz E, Brezis M, Rosenmann E, et al. Anti dsDNA antibodies bind directly to renal antigens and induce kidney dysfunction in the isolated perforated kidney. J Immunol. 1989;142:3076–3082.
11 Radic MZ, Weigert M. Genetic and structural evidence for antigen selection of anti-DNA antibodies. Annu Rev Immunol. 1994;12:487–520.
12 Rahman A, Giles I, Haley J, et al. Systematic analysis of sequences of anti-DNA antibodies—relevance to theories of origin and pathogenicity. Lupus. 2002;11:807–823.
13 Martensen ES, Fenton KA, Rekring OP. Lupus nephritis—the control role of nucleosomes revisited. Am J Pathol. 2008;172:275–283.
14 Munoz LE, Gaipl US, Franz S, et al. SLE—a disease of clearance deficiency? Rheumatology (Oxford). 2005;44:1101–1107.
15 van Bavel CC, Fenton KA, Rekvig OP, et al. Glomerular targets of nephritogenic autoantibodies in systemic lupus erythematosus. Arthritis Rheum. 2008;58:1892–1899.
16 Kalaaji M, Sturfelt G, Mjelle JE, et al. Critical comparative analyses of anti-alpha-actinin and glomerulus-bound antibodies in human and murine lupus nephritis. Arthritis Rheum. 2006;54:914–926.
17 Amoura Z, Koutouzov S, Chabre H, et al. Presence of antinucleosome autoantibodies in a restricted set of connective tissue diseases: antinucleosome antibodies of the IgG3 subclass are markers of renal pathogenicity in systemic lupus erythematosus. Arthritis Rheum. 2000;43:76–84.
18 Gioud M, Kaci MA, Monier JC. Histone antibodies in systemic lupus erythematosus. A possible diagnostic tool. Arthritis Rheum. 1982;25:407–413.
19 Cocca BA, Seal SN, D’Agnillo P, et al. Structural basis for autoantibody recognition of phosphatidylserine-beta 2 glycoprotein I and apoptotic cells. Proc Natl Acad Sci U S A. 2001;98:13826–13831.
20 Wellmann U, Letz M, Herrmann M, et al. The evolution of human anti-double-stranded DNA autoantibodies. Proc Natl Acad Sci U S A. 2005;102:9258–9263.
21 Bootsma H, Spronk P, Derksen R, et al. Prevention of relapses in systemic lupus erythematosus. Lancet. 1995;345:1595–1599.
22 Tseng C, Buyon JP, Kim M, et al. The effect of moderate-dose corticosteroids in preventing severe flares in patients with serologically active, but clinically stable, systemic lupus erythematosus. Arthritis Rheum. 2006;54:3623–3632.
23 Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62:222–233.
24 Navarra SV, Guzman RM, Gallacher AE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377:721–731.
25 Fava C, Isenberg D. B cell depletion therapy in SLE—what are the current prospects for its acceptance. Nat Rev Rheum. 2009;5:711–716.
26 Alarcon-Segovia D, Tumlin JA, Furie RA, et al. LJP 394 for the prevention of renal flare in patients with systemic lupus erythematosus: results from a randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2003;48:442–454.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


