Chapter 27 Cardiopulmonary Disease in SLE
Cardiopulmonary Manifestations
The heart and the lungs can frequently be affected during the course of systemic lupus erythematosus (SLE), because of either the disease itself or the unwanted effects of lupus therapies (Boxes 27-1 and 27-2). The true prevalence of involvement of both systems is unknown, given the protean clinical presentation and the lack of well-designed epidemiologic and/or necropsy studies. In a study of 90 necropsies in Argentina, some degree of pulmonary involvement was found in almost 99% of patients, with pleural disease and infections being the most common.1 However, clinically apparent disease is much less frequent in large observational cohorts.2
Box 27-1
Respiratory Involvement in Systemic Lupus Erythematosus
Cardiovascular disease is one of the main prognostic predictors in SLE.2 Patients with lupus are prone to the development of early atherosclerosis, which is specifically covered in another chapter of this book. In addition, the hearts of patients with lupus can be involved in other ways, from the pericardium to the endocardium.3
Serositis: Pleurisy and Pericarditis
Pleurisy and pericarditis are the most frequent pulmonary and cardiac manifestations of SLE. The Euro-Lupus (European Working Party on Systemic Lupus Erythematosus) observational cohort found a prevalence of serositis at disease onset of 17% with a cumulative incidence of 36%.2 Pleuritic chest pain may occur during the course of lupus in up to 60% of patients.4
The clinical features of lupus serositis are indistinguishable from those of pleurisy and pericarditis from other causes. Pleuritic chest pain may be unilateral or bilateral and is usually located at the costophrenic margins, either anterior or posterior. Attacks of pleuritic pain often last for several days and may persist for weeks, often accompanied by cough and dyspnea. Pericarditis usually manifests as precordial pain aggravated by deep breathing and decubitus, typically improving with sitting up. Fever and other clinical signs of lupus activity, such as arthralgia/arthritis and rashes, are common. On physical examination, friction rubs may be heard. A decreased intensity of cardiac and respiratory sounds is typical in the presence of large effusions. Respiratory compromise and pericardial tamponade are distinctly unusual in lupus serositis.3,4
Pleural and pericardial effusions may or may not occur during the course of serositis, so their presence is not necessary for the diagnosis, which is often based on clinical grounds. They can be apparent in radiographic studies, affecting one or both sides of the pleural cavity with variable intensity (Figure 27-1) and/or showing as an enlarged cardiac silhouette if a significant amount of pericardial fluid is present. Computed tomography (CT) scan and echocardiography have higher sensitivity to detect small effusions (Figure 27-2). Electrocardiographic findings in pericarditis include PR segment depression and widespread ST segment elevation.
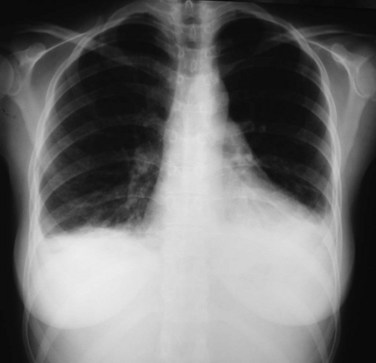
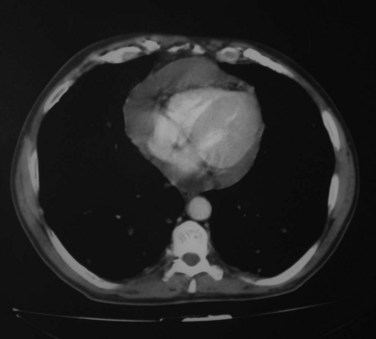
Analysis of the pleural fluid is frequently required to complete the diagnosis. Pericardiocentesis, on the other hand, is a complex invasive procedure, so it is most often not performed. The pleural fluid in SLE is almost always exudative. Both lymphocyte and polymorphonuclear predominance can be seen. In most patients with lupus pleuritis, the pleural fluid glucose concentration is greater than 60 mg/dL. This contrasts with the finding of low glucose levels in the pleural fluid of patients with rheumatoid pleurisy, in whom the glucose concentration is less than 30 mg/dL in 75% of cases.5 Low pleural fluid glucose concentrations may also occur in malignant effusions, empyema, and tuberculosis.5
The presence of antinuclear antibodies (ANAs) in the pleural fluid may be a useful diagnostic test for lupus. Most investigators agree that the negative predictive value of ANAs in pleural fluid is very high for SLE, that is, an ANA-negative pleural effusion is very unlikely to be due to lupus, even in a patient with known SLE, in whom alternative causes should be pursued.6–8 On the other hand, ANA-positive pleural fluid can be found in patients without lupus. In those cases, malignancy should be put first in the list of possible causes.7,8 The serum-to–pleural fluid ANA ratio does not seem to improve the performance of ANA testing at titers over 1 : 160; thus the use of this ratio is not recommended in clinical practice.7,8 Limited data point to similar performance of ANA testing in pericardial fluid.7
Acute Lupus Pneumonitis
Acute lupus pneumonitis is a rare manifestation of lupus, with an incidence ranging between 1% and 12%.9 Unfortunately, in more than 50% of cases, acute lupus pneumonitis is the presenting manifestation of SLE, making early diagnosis and treatment more difficult.10
The histopathology of the lung in acute lupus pneumonitis has been examined in a few patients. Alveolar hyaline membranes and persistent cell infiltrates have been found. Other findings at the autopsy included acute alveolitis, interstitial edema, and arteriolar thrombosis. Granular deposits of immunoglobulin (Ig)G, complement component C3, and DNA have been seen in the alveolar septa of patients with acute lupus pneumonitis.10
Patients with acute lupus pneumonitis usually present with fever, dyspnea, cough productive of scanty sputum, tachypnea, and pleuritic chest pain.9 Physical findings commonly include basal crackles, and, when severe, central cyanosis may be present. Chest radiographs and CT scans show diffuse alveolar infiltrates with a predilection for the bases in all patients (Figure 27-3). Pleural effusion can be present in up to 50% of patients.10 Respiratory insufficiency is the rule, with many patients having a fulminant course. Adult respiratory distress syndrome may occur with acute lupus pneumonitis, greatly increasing mortality. Anti-DNA antibodies are usually present along with other data revealing lupus activity. Anti-Ro antibodies are also frequently found.10
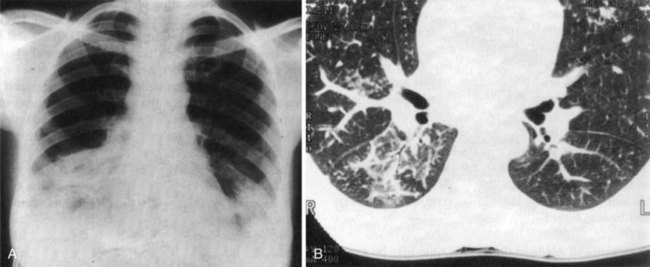
Acute lupus pneumonitis is clinically similar to alveolar hemorrhage (see later). The differential diagnosis should always include pulmonary infections; thus, bronchoalveolar lavage, including specific cultures of the fluid, can be very useful in this setting.9 Acute lupus pneumonitis should be suspected in patients with known SLE and an acute pulmonary picture in the context of high disease activity. Tests for ANAs, anti-DNA antibodies, and complement levels as well as a search for specific clinical manifestations of lupus should be included in the diagnostic routine for young patients presenting with fever and alveolar infiltrates of noninfectious origin.
Owing in part to the frequent delay in the diagnosis, the mortality rate for acute lupus pneumonitis may approach 50%, although prompt and aggressive therapy may substantially improve the prognosis.9
Pulmonary Hemorrhage
Pulmonary hemorrhage is a rare, devastating, and frequently fatal manifestation of SLE.4 The histopathology of the lung is nonspecific, showing diffuse, intraalveolar hemorrhage with intact erythrocytes, and hemosiderin-laden macrophages in the alveoli. Pulmonary capillaritis can be seen in up to 80% of lung tissue specimens, although its presence varies among different series.11
The clinical presentation of alveolar hemorrhage closely resembles that of acute lupus pneumonitis. Fever, dyspnea, and cough are common presenting features. Blood-stained sputum and, eventually, frank hemoptysis can appear in more than 50% of patients, so their absence does not exclude the diagnosis. The clinical course is rapidly progressive over hours or days, with increasing tachypnea, arterial hypoxemia, tachycardia, and acute respiratory distress. The hemoglobin concentration usually drops suddenly, and chest radiographs show bilateral pulmonary infiltrates, with a predominantly alveolar pattern, often extending to the bases but occasionally unilateral in distribution. CT may help confirm radiographic findings and exclude alternative conditions such as infection and cancer. In the absence of hemoptysis, a rapidly falling hemoglobin and diffuse lung infiltrates should alert the clinician to the possibility of lung hemorrhage. Single-breath diffusing capacity for carbon monoxide (CO) is typically raised owing to the presence of blood in the alveolar spaces, although most patients are too ill to undergo this investigation.12 Bronchoalveolar lavages are almost universally hemorrhagic, with the presence of hemosiderin-laden macrophages as an indirect sign of alveolar bleeding.11,12 Open lung biopsy is not generally needed to establish the diagnosis.
Pulmonary hemorrhage is not a common presenting feature of SLE. In a series from Mexico, one third of patients with lupus and alveolar hemorrhage had no previous diagnosis of SLE.13 Interestingly, most other researchers report that more than 80% of patients with pulmonary hemorrhage have known lupus, usually of recent diagnosis.11,12 In fact, multisystem disease is almost the rule, with renal and neurologic disease as the most frequent accompanying features.11,13
The prognosis of massive pulmonary hemorrhage in patients with SLE is grave, despite aggressive treatment, with mortality rates exceeding 50% in most published series.11–14 The presence of thrombocytopenia, renal failure, and infection and the need for mechanical ventilation have all been identified as adverse prognostic factors.11–13
Chronic Diffuse Interstitial Lung Disease
Diffuse interstitial lung disease (ILD) is a well-recognized pulmonary manifestation of systemic autoimmune diseases, particularly systemic sclerosis and dermatomyositis; however, it is much less common in SLE. The prevalence of symptomatic ILD in SLE has been calculated to be approximately 3% to 8%, being more frequent as disease duration increases.15
The classification of idiopathic interstitial pneumonias has been updated.16 According to this classification, nonspecific interstitial pneumonia (NSIP) is the most common histologic pattern found in patients with SLE.15 Lung biopsies show interstitial inflammation, fibrosis, or a combination of the two.16 Usual interstitial pneumonia (UIP) shows a fibrotic pattern, whose more characteristic associated condition is idiopathic pulmonary fibrosis, although UIP can appear in connective tissue diseases, including SLE.16 Other patterns are lymphocytic interstitial pneumonia (LIP), most common in Sjögren syndrome but also seen in a few patients with SLE, and organizing pneumonia15 (for the latter, see later discussion of airway obstruction).
The presentation of ILD in SLE resembles that of lung disease in systemic sclerosis and rheumatoid arthritis. ILD can occur at any time during the course of SLE, but in most cases it develops in patients with long-standing disease. The most common clinical manifestations are slowly progressing dyspnea on exertion and nonproductive cough. Inspiratory crackles are evident as disease advances. Other lupus features may be present as well in variable combinations. An association of anti-Ro and anti–U1 ribonucleoprotein (anti–U1-RNP) antibodies with ILD has been suggested by some authorities, although this relationship is disputed.4
Chest radiographic findings may range from almost normal to severe honeycombing, most frequently showing reticulonodular patterns.16 Lower lobe disease is the usual finding. The common pattern of nonspecific interstitial pneumonia on CT is a ground-glass appearance, whereas UIP manifests as a reticulonodular infiltrate with variable fibrosis along with parenchymal destruction and volume loss (Figure 27-4).16 Pulmonary function tests typically show a restrictive pattern with a reduced diffusing capacity for CO.9 A lung biopsy is not always necessary to establish the diagnosis, but it may be indicated to exclude infection or cancer in selected cases.
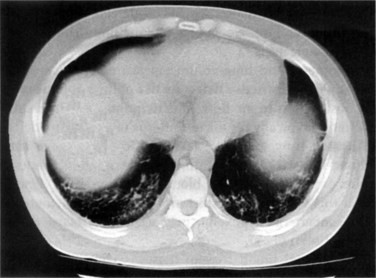
FIGURE 27-4 High-resolution CT chest scan showing bilateral basal interstitial fibrosis with honeycombing.
Pulmonary Embolism
Pulmonary embolism should always be considered in the setting of chest pain, dyspnea, and respiratory insufficiency, especially if antiphospholipid antibodies are present. Spiral CT scan should be performed in cases of clinical suspicion.17 The lungs are commonly involved in catastrophic antiphospholipid syndrome, with the usual presentation of acute respiratory distress syndrome. Bucciarelli reported pulmonary involvement in 150 of 220 (68%) patients with catastrophic antiphospholipid syndrome; 47 (21%) patients were diagnosed as having acute respiratory distress syndrome. Prognosis was poor, and 19 (40%) of these patients died. Histologic analysis showed that 7 of 10 patients had a thrombotic microangiopathy (see Chapter 42 for a further discussion of thrombosis and antiphospholipid antibodies).18
Reversible Hypoxemia
A relatively rare syndrome of acute reversible hypoxemia in acutely ill patients with SLE without evidence of parenchymal lung involvement may occur.19 Although some patients have mild pleuropulmonary symptoms, chest radiographic and CT findings are typically normal. Patients present with hypoxemia and hypocapnia with a wide alveolar-arterial gradient. The pathogenesis of the syndrome is unclear. A relationship with disease activity has been noted.20 Complement activation may lead to diffuse pulmonary injury with leukocyte–endothelial cell adhesion and leukoocclusive vasculopathy within pulmonary capillaries.17 Indeed, most cases respond to immunosuppression, with rapid improvement of gas exchange within a few days.17
Pulmonary Hypertension
Pulmonary hypertension (PH) is characterized by the progressive increase in pulmonary vascular resistance eventually leading to right ventricular failure and premature death. It is defined as a resting mean pulmonary arterial pressure (mPAP) higher than 25 mm Hg measured by right heart catheterization.21 According to the latest classification of PH—now known as the Dana Point classification, from the location of the 4th World Symposium on Pulmonary Hypertension, at Dana Point, California, in 2008 at which the update was adopted—this condition can be secondary to a number of disorders, including pulmonary diseases and/or hypoxemia, left heart disease, thromboembolic disease of the pulmonary arteries, and a miscellanea of other causes, such as sarcoidosis and lymphangiomyomatosis.22 For the diagnosis of the subclass known as pulmonary arterial hypertension (PAH), a pulmonary arterial wedge pressure lower than15 mm Hg is required.22
PAH associated with systemic autoimmune diseases is included within group I of the Dana Point classification. PAH is a recognized complication of this group of diseases, particularly systemic sclerosis and, to a much lesser degree, SLE, mixed connective tissue disease, inflammatory myopathies, and Sjögren syndrome.22 In addition, PH may be secondary to other complications, such as interstitial lung disease, valvular heart disease, and pulmonary thromboembolism, that may occur during the course of lupus.
Plexiform lesions of the pulmonary arteries are the hallmark of PAH, whether or not it is secondary to SLE or other connective tissue disorders.1 Medial hypertrophy and intimal fibrosis of the branches of the pulmonary artery may be seen. Thrombosis and vasculitis have also been reported in a few patients.9
The pathogenesis of PAH in SLE is likely to be multifactorial. Several factors have been implicated, such as recurrent vasospasm, vasculitis, and thrombotic vascular occlusion. In addition, PH can be secondary to pulmonary fibrosis, chronic thromboembolic disease, and left ventricular dysfunction. Increased levels of endothelin-1 have been proposed as a possible mechanism for PAH in patients with lupus.23 A number of clinical and immunologic variables have been reported as potential markers of an increased risk for PAH: Raynaud phenomenon,24,25 disease activity,26,27 and presence of antiphospholipid antibodies28,29 and anti–U1-RNP antibodies.27,30
The actual prevalence of PH in patients with lupus is unknown, ranging from less than 0.5% to 14%,23 depending on the series. Such discordant results actually reflect the varying definitions of PH. In some series, the diagnosis has been established following a single echocardiographic calculation of systolic pulmonary arterial pressure, with a cutoff point between 30 and 40 mm Hg, depending on the study. This diagnostic strategy, which does not fulfill current recommendations,21 may well overestimate PH prevalence and also bias the identification of clinical and immunologic predictors. In a necropsy series of 90 patients with SLE, histologic evidence of PAH was found in 4%.1
The symptoms of PAH in SLE are nonspecific and similar to those of patients with other forms of this condition. The most common complaints are dyspnea on exertion, chest pain, and chronic nonproductive cough.9 Weakness, palpitations, edema, and/or ascites may also gradually occur as disease progresses and the right ventricle becomes involved. The physical findings include a loud second pulmonary heart sound, systolic murmurs, and right ventricular lift. Chest radiographic findings include prominent pulmonary arteries and clear lung fields, with cardiomegaly in more advanced cases. Electrocardiography may show changes of right ventricular hypertrophy. Pulmonary function tests in PAH typically show a diminished CO diffusion with normal lung volumes. A restrictive pattern is seen in PH secondary to lung fibrosis. Echocardiogram is considered the best screening test for PH. Calculated PAP values exceeding 40 mm Hg warrant further investigations.31 Cardiac catheterization is the definitive diagnostic test for PAH, demonstrating the elevation of the mean pulmonary artery pressure to more than 25 mm Hg at rest with a normal wedge pressure, without evidence of intracardiac or extracardiac shunting. For therapeutic purposes, a vasodilator test would be included in the procedure, because a positive response identifies those few cases that can respond to calcium-antagonist drugs (see section on treatment).
PAH has been identified as a predictor of morbidity and mortality in SLE.2,24 Cardiac failure and sudden death, presumably due to arrhythmias, are the most common causes of death. The survival of patients with lupus and PAH has been considered poor.9 However, a British national registry of PH starting in 2001 has shown 1- and 3-year survival rates of 78% and 74%, respectively, for PAH secondary to SLE, significantly higher than those of systemic sclerosis–associated PAH.32 These figures may reflect the availability of new, effective therapies for PAH, but also the differential, specific characteristics of SLE-associated PAH.
Shrinking Lung Syndrome
Shrinking lung syndrome refers to a condition typical of SLE that consists of a purely restrictive respiratory disease with normal lung parenchyma and markedly decreased lung volumes usually evident in radiographic studies, which show elevated hemidiaphragms and basal atelectasis (Figure 27-5).
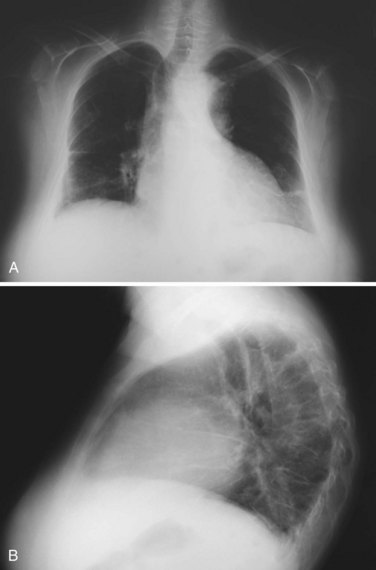
Diaphragmatic dysfunction has been advocated as the main pathogenetic mechanism of shrinking lung syndrome.33 However, Laroche found no evidence of isolated weakness of the diaphragm in 12 patients with SLE and this syndrome.34 Evidence of chronic pleural disease has not been demonstrated either.33 Anti-Ro antibody positivity has been also linked with the occurrence of shrinking lung syndrome.35
Shrinking lung syndrome usually manifests as exertional dyspnea of variable severity, which can progress over a period of weeks or months. Orthopnea may also occur, attributed to diaphragmatic weakness.33 Pleuritic chest pain is reported frequently, and a previous history of pleurisy and pericarditis is common.36 Anti-Ro antibodies may be present, although they do not offer an additional diagnostic aid. Physical examination is remarkably normal.
Chest radiography typically shows elevated hemidiaphragms, although this is not a universal finding and its absence does not exclude the diagnosis.33 Pleural effusions, pleural thickening, and atelectasis may be also evident on plain films or CT scans. Pulmonary function tests show a marked restrictive pattern, with decreased forced vital capacity (FVC). Carbon monoxide diffusion corrected by lung volumes is typically normal.
The prognosis of this syndrome is usually good, with most patients showing long-term stabilization.33
Airway Obstruction
Airway obstruction can be found in a substantial proportion of patients with lupus, up to 20% depending on the series, but severe forms are rare in the absence of other concomitant causes such as smoking.4 Specific, lupus-related obstructive airway disease is much more uncommon and is caused mainly by bronchiolitis.33
Bronchioles are small airways that do not have cartilage in their walls. The term bronchiolitis is applied to a variety of inflammatory disorders involving the bronchioles.37 Two types of primary bronchiolitis can be seen in patients with lupus. The first one is constrictive bronchiolitis, also known as obliterative bronchiolitis or bronchiolitis obliterans.38 The distinctive pathologic pattern of constrictive bronchiolitis consists of peribronchiolar fibrosis, which surrounds the lumen, resulting in extrinsic compression and obliteration of the airway. Lupus is a rare cause of this type of bronchiolitis, which is most often idiopathic or due to drugs—such as penicillamine—inhalation injury, chronic transplant rejection, or, among autoimmune disorders, rheumatoid arthritis.38 The clinical picture is that of cough and progressive dyspnea. Physical examination may reveal wheezing and inspiratory crackles. Chest radiographic findings are distinctively normal. On the other hand, CT shows a pattern of mosaic attenuation, with heterogeneous lung density due to decreased perfusion of areas with bronchiolar obstruction and blood flow redistribution to normal areas. Pulmonary function tests typically show a nonreversible obstructive pattern, with predominant involvement of distal airways.39
The second type of bronchiolitis that can be seen in patients with lupus is bronchiolitis obliterans organizing pneumonia, also known as cryptogenetic organizing pneumonia.16 Actually, bronchiolar involvement is not predominant in this entity, which consists of universal polypoid intraluminal plugs of proliferating fibroblasts and myofibroblasts within alveolar ducts and spaces, and occasional signs of organization within the bronchioles.16 This makes a major difference with obliterative bronchiolitis, in which the fibrosing reaction is peribronchiolar.38
Clinical findings include cough and dyspnea of acute/subacute onset, often with prominent systemic symptoms such as fever, myalgia, and weight loss. Occasionally, the clinical presentation may resemble adult respiratory distress syndrome.38 Chest radiography and CT demonstrate prominent alveolar consolidation with bronchogram. Lung function tests show a restrictive ventilatory pattern with usually moderately reduced CO diffusion. Airflow obstruction is present in a few patients.16
The prognosis of these two conditions is also radically different. Although organizing pneumonia usually responds well to immunosuppressive therapy, constrictive bronchiolitis is often a gradually worsening disease with a high mortality rate within a few months.38

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


