Chapter 48 Systemic Glucocorticoid Therapy in SLE
Endogenous and Synthetic Glucocorticoids
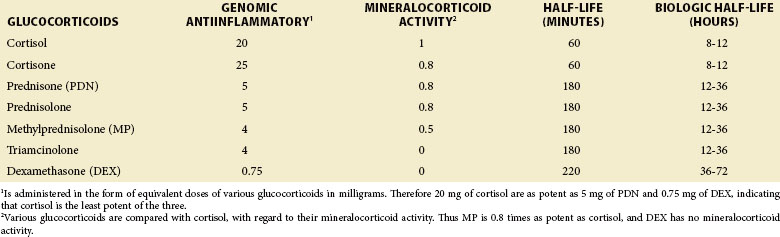
Synthetic GCs, more potent and with fewer MC effects than cortisol, have been developed. The biochemical structure of cortisol and synthetic GC is shown in Figure 48-1 and their pharmacologic properties are compared in Table 48-1. Regulatory mechanisms of synthetic GCs, as they apply to binding to the corticosteroid-binding globulin (CBG), tissue-specific metabolism, affinity for GC receptors (GRs), and interaction with transcription factors, may substantially differ from those of native GCs.
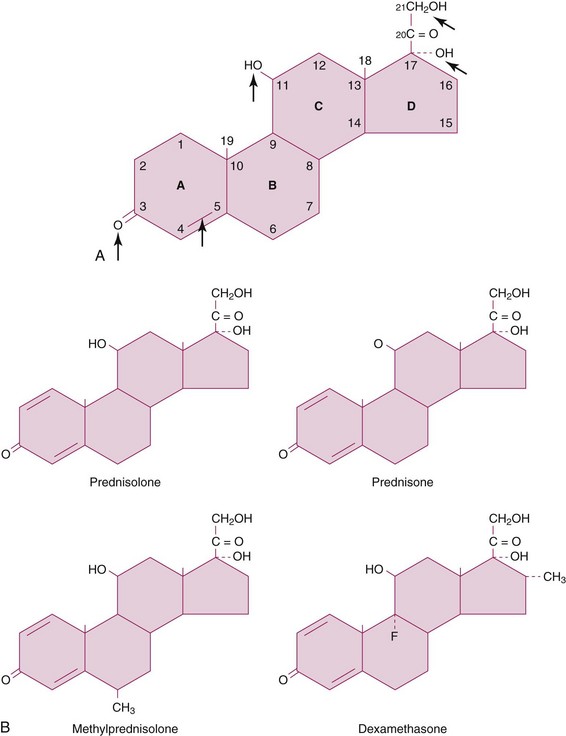
FIGURE 48-1 A, Structure of cortisol (hydrocortisone). All Δ4 double-bond groups, 3-keto group, and 11β-OH groups are essential for glucocorticoid (GC) function, and the first two groups are also required for mineralocorticoid (MC) activity. The hydroxyl group at C21 is required for MC activity and is present on all natural and synthetic GCs. The 17α-hydroxyl group, present on cortisol and synthetic GCs (but not on corticosterone), enhances GC potency. B, Structure of selected common synthetic GCs. The addition of a Δ1 double bond on cortisol (as in all shown GCs) selectively increases GC activity and delays GC metabolism. The methyl group at position 6α (methylprednisolone [MP]) increases GC over MC activity even further. Notably, fluorination at the 9α position (fludrocortisone; not shown) greatly enhances GC and MC activity (the latter significantly more than the former). However, when modified with a Δ1 double-bond group and a methyl group substitution at C16α, fludrocortisone loses all MC activity and becomes dexamethasone (DEX).
The great need for improved synthetic GCs with less adverse effects and intact antiinflammatory and immunosuppressive actions has led to the development of newer synthetic GCs by modifying their pharmacokinetic or pharmacodynamic properties. Budesonide is an example of a GC with high topical activity but low systemic bioavailability as a result of rapid first-pass liver inactivation and has been used by inhalation in asthma and orally in Crohn disease. Of more interest to rheumatology is the development of liposomal formulations of GCs. These agents, which have been successfully used in animal models of arthritis, preferentially target macrophages in tissues, such as the synovium and the spleen, and achieve antiinflammatory effects without the need for repeated administration.1,2 More recently, modified-release prednisone (PDN) tablets have been designed that release PDN 4 hours after their ingestion at bedtime and before the secretion of interleukin (IL)-6, which normally peaks at approximately 8:00 AM. This PDN chronotherapy was tested with favorable results against regular PDN therapy in patients with rheumatoid arthritis (RA) in a randomized controlled trial (RCT) with open-label extension.3
Yet another strategy proposed for enhanced GC effect with less adverse effects is combination therapy with other drugs that work synergistically or additively. Examples include β-adrenergic agents, phosphodiesterase inhibitors, and nitric oxide–conjugated GCs (nitrosteroids), especially in the treatment of asthma and chronic obstructive pulmonary disease (COPD).4–6
Molecular Mechanisms of Glucocorticoid Action
GC effects are mainly mediated via specific GRs in the cytoplasm that operate as hormone-activated transcriptional regulators.7 Hydrocortisone and other GCs are also capable of binding MC receptors (MRs) with higher affinity than they bind GRs and mediating aldosterone-like effects (see Table 48-1). GR specificity, at relatively low baseline body cortisol levels, is maintained because of the action of 11 beta–hydroxysteroid dehydrogenase enzyme 2 (11β-HSD2), a steroid metabolizing enzyme expressed at MC-sensitive tissues (e.g., the kidney) that metabolizes hydrocortisone to the inactive cortisone.
GRs, when inactive, are bound to several receptor-associated proteins, such as the heat shock protein (HSP) 90.7,8 Upon GC binding, GRs dissociate from these proteins and translocate to the nucleus. There they mediate their effects mainly via either GR homodimerization and direct transactivation/transrepression of genes or indirectly via protein-protein interactions with other transcription factors (Figure 48-2). Indirect transrepression is thought to mediate antiinflammatory effects within a few hours, and direct transactivation/transrepression to mediate both adverse effects and antiinflammatory effects within days. This concept has led to the development of synthetic selective glucocorticoid receptor agonists (SEGRAs) with dissociated activity for indirect transrepression (potent) and direct transactivation (weak) and their successful use in an animal model of skin inflammation.9 Notably, GCs, by inducing mitogen-activated protein kinase (MAPK) phosphatase 1 (MKP1) and inhibiting p38 MAPK, can downregulate proinflammatory genes, such as cytokines, cyclooxygenase (COX)-2, and inducible nitric oxide synthase (iNOS), as well as via posttranscriptional mechanisms.6
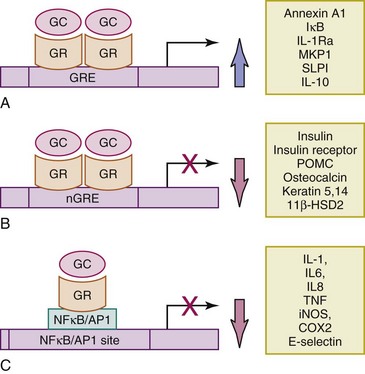
FIGURE 48-2 Mechanisms of genomic effects of GCs. A, Direct transactivation of genes through binding of activated GR dimers to GRE on the corresponding gene promoter or enhancer regions. Examples of such transactivated antiinflammatory genes include ANXA1, also known as lipocortin 1, and its receptor; FPR2 or ALXR; IκB, an inhibitor of NF-κB; IL-1Ra, an inhibitor of IL-1β; MAPK phosphatase 1 (MKP1) or DUSP1; SLPI; SLAP 1; neutral endopeptidase; and IL-10. Examples also include enzymes of gluconeogenesis, responsible for hyperglycemic effects of GC7 and GC8. B, Direct transrepression of genes (often responsible for GC adverse effects) through binding of activated GR dimers to nGRE on the corresponding gene promoter or enhancer reGC-induced osteonecrosis.95 Examples include insulin precursor, insulin receptor, POMC, osteocalcin, keratins 5 and 14, cyclin D1, and 11β-HSD2. C, Indirect transrepression through protein-to-protein cross-talk; activated GR monomers interact and inhibit proinflammatory transcription factors, such as NF-κB and AP-1 11, 13. Examples include the following: (1) Cytokines: IL-1, IL-2, IL-4, IL-5, IL-6, IL-12, TNF, and GMCSF, among others. (2) Chemokines: IL-8, MCP-1, MIP-1α, and eotaxin, among others. (3) Proinflammatory enzymes: inducible nitric oxide synthase (iNOS), COX2, and collagenase. (4) Adhesion molecules: ICAM-1 and E-selectin, among others.
Besides having GR-mediated transcriptional genomic effects that depend on new protein synthesis and therefore have a delayed onset (at least 30 minutes and usually several hours), GCs may have more rapid (seconds or minutes) nongenomic effects.10–13 These GC effects usually occur at relatively large pharmacologic or pulse-GC doses and are mediated by either membranous or cytosolic GRs. Examples include the rapid dissociation of T-cell receptor–associated lymphocyte-specific protein tyrosine kinase (Lck) and Fyn (a Src-family tyrosine kinase) and therefore the inhibition of T-cell signaling.8 Moreover, GCs, especially at pulse-GC doses, may cause nonspecific physicochemical interactions with cellular membranes and cause immunosuppression by inhibiting calcium and sodium cycling across the plasma membrane of immune cells.12 A recent study showed that novel GCs, conjugated to glycine or lysine to block genomic effects, were able to rapidly inhibit neutrophil degranulation and immunoglobulin E (IgE)–mediated histamine release from mast cells.11
Antiinflammatory and Immunosuppressive Effects
The biologic effects of GCs are multiple; they affect all tissues and are essential for body homeostasis during normal or stress conditions. Although GCs are used to suppress inflammation and pathologic immune responses in clinical medicine, a growing number of studies paradoxically attribute immune-enhancing effects to these agents. It seems that endogenous GCs have an important overall regulatory role in modulating immune responses that develop to such stressors as infections.13 For example, GCs, on the one hand, act permissively to help immune responses develop adequately and in a timely fashion to fight the invading organisms, and yet, on the other hand, they act suppressively to restrain a potentially deleterious overshoot of these same responses. Parameters that determine the direction of GC-immune effects primarily include the serum levels and timing of GC exposure relative to the initiation of stress. Higher (pharmacologic) levels such as those occurring after the initiation of stress are, in general, immunosuppressive, whereas lower (physiologic) levels of GCs present before stress initiation may enhance immune responses. Notably, acute stresses (or a short exposure to GCs) enhance immune responses, whereas long-term exposure to stress or to GCs has the opposite effect.
The antiinflammatory effects of GCs, as shown in Figure 48-2 and Box 48-1, are complex. At the molecular level, GCs act on various cells (e.g., neutrophils, monocytes-macrophages, fibroblasts, endothelial cells) by both genomic and nongenomic effects to inhibit the synthesis and secretion of inflammatory mediators, as well as promote the synthesis and secretion of antiinflammatory proteins (see Figure 48-2). At the cellular level, they not only inhibit the initiation of acute inflammation with the blockade of small-vessel vasodilation and leukocyte (polymorphonuclear neutrophil [PMN] and eosinophil) migration to tissues in response to damage signals, but they also facilitate the nonphlogistic disposal of inflammatory cells and the resolution of inflammation.14,15
Box 48-1
Important Antiinflammatory and Immunosuppressive1 Effects of Glucocorticoids on Various Cells of the Immune System4,7,9,76,77
Glucocorticoid Resistance
Pharmacokinetic causes of resistance to GCs may include impaired oral bioavailability as a result of decreased GC absorption (e.g., by cholestyramine) or increased GC metabolism (e.g., by drugs such as barbiturates). With regard to pharmacodynamic causes, tissue-specific GC resistance has been studied best in steroid-resistant bronchial asthma, in which the lack of GC benefits for airway inflammation contrasts with a high incidence of GC adverse effects from other organs. Cytokines, secreted in the context of such diseases as bronchial asthma, RA, SLE, and depression, are thought to play an important role in mediating tissue-specific GC resistance by inhibiting GR function.16 In addition and more relevant to patients with SLE, a recent study has shown that signaling via Toll-like receptors (TLRs) 7 and 9 blocks the inhibitory effect of GCs on plasmacytoid dendritic cells (pDCs) with regard to type I interferon (IFN) production by these cells.17 The authors of the study have proposed that therapy with TLR 7 and 9 inhibitors could work as GC-sparing agents in patients with SLE. Finally, high levels of P-glycoprotein (P-gp) have been noted in lymphocytes of patients with active lupus that was resistant to high doses of prednisolone.18 GC resistance was reversed after intensive immunosuppressive therapy and/or cyclosporine, which functions as a competitive inhibitor of P-gp.
Pharmacokinetics and Drug Interactions
Oral absorption of GCs is excellent whether on an empty or full stomach. Once in the circulation, a large fraction of GCs (90% for hydrocortisone) binds to serum proteins. Only their free fraction is biologically active. Of the two GC-binding proteins, transcortin or CBG binds to GCs with high affinity and low capacity, whereas albumin binds with low affinity and high capacity. Although hydrocortisone and prednisolone bind to both CBG and albumin, their protein binding is concentration dependent and varies from 90% at lower doses (i.e., with standard oral doses) to 60% at higher doses. In contrast, methylprednisolone (MP) and dexamethasone (DEX) bind almost exclusively (99%) to the high-capacity albumin and therefore have concentration-independent protein-bound fractions (60% to 70%). The 11-keto GC derivatives such as PDN and cortisone are inactive unless reduced by 11-β-HSD1 in the liver to their 11-OH analogs, prednisolone and hydrocortisone (see Figure 48-1). Inactivation of GCs occurs predominantly in the liver and involves the sequential reduction of the Δ4 double bond (i.e., the rate-limiting step in cortisol metabolism), and the 3-keto group (see Figure 48-1). Glucuronidation and sulfation follow, which confer water solubility and allow for urine excretion. Additionally, 6β-hydroxylation by the cytochrome P450 microsomal enzyme (family 3, subfamily A, polypeptide 4) (CYP3A4) also enhances water solubility and urinary excretion of GC. Serum half-lives of different GCs vary from 60 to 300 minutes. However, biologic half-lives of GCs are dependent on their tissue levels and are much longer than their serum half-lives (see Table 48-1). In addition to the previously mentioned inhibition of enteric GC absorption by cholestyramine, other important drug interactions also exist. Drugs that induce hepatic microsomal enzymes (especially CYP3A4), such as phenobarbital, phenytoin, rifampin, and carbamazepine, increase GC elimination. In contrast, CYP3A4 inhibitors, such as ketoconazole and clarithromycin, increase GC activity.
General Principles of Glucocorticoid Therapy
Uncontrolled disease activity in patients with SLE can be both debilitating and life threatening and thus demands rapid and effective intervention. The value of therapy with high doses of GCs in such a setting (e.g., in diffuse proliferative glomerulonephritis [DPGN]) is unquestionable. On the other hand, GCs can have multiple complications that are directly related to the dose and duration of therapy. (See section under “Adverse Effects of Glucocorticoids” later in this chapter.) In fact, Sergent and colleagues19 have shown increased infection-related mortality in patients with severe neuropsychiatric SLE (NP-SLE) when treated with PDN doses of more than 100 mg/day for an average of 37 days (range, 8 to 68 days).19 On the other hand, low-dose GC therapy (i.e., a dose equivalent of 7.5 mg or less of PDN per day) appears to be tolerated better but is not free of risks; complications such as growth suppression, osteoporosis (OP), and cataract formation can still occur. Therefore the ultimate goal of therapy should always be the complete cessation of GCs, if possible.
The First European Workshop on GC therapy proposed a standardized nomenclature for GC doses and GC treatment regimens, taking into account the percent saturation of GRs at different doses and clinical practice.20 The nongenomic effects, which become increasingly important with very high–dose GC and pulse-GC therapies, were also noted. These definitions have been adopted in this text (Table 48-2). The most effective approach to initiating high-dose or very high–dose GC therapy for severe SLE disease, especially when constitutional symptoms (e.g., high fever, prostration) are present, is to administer it in two to four doses per day. A notable exception is the management of severe focal proliferative glomerulonephritis (FPGN) or DPGN, in which once-a-day regimen is adequate. Should the condition prove GC unresponsive, then the use of pulse-GC or additional immunosuppressive agents or both is necessary. Most disease complications will respond in less than 1 to 2 weeks. However, markers of lupus nephritis (especially proteinuria) may take more than 2 to 6 weeks to improve.
TABLE 48-2 Usual Regimens of Systemic Glucocorticoid Therapy in Patients with Systemic Lupus Erythematosus1









