CHAPTER 32 Spinal Disorders Associated with Skeletal Dysplasias and Metabolic Diseases
Spinal disorders associated with skeletal dysplasias and metabolic diseases may be called orthopaedic trivia by some. None of these conditions are found in a large number of patients, and the numbers of syndromes and subclassifications seem to be almost endless. More than 200 types of short stature syndromes have been described to date, and the mapping of the human genome has led to discovery and clarification of many new metabolic disorders. Despite the relative rarity of these conditions in any spine surgeon’s practice, it is important for physicians and surgeons to know enough features of these disorders to allow for their early recognition and diagnosis. Because the natural history of spinal disorders in each of these syndromes is often quite different, it is important initially to establish as accurate a diagnosis as possible, particularly in disorders in which spinal deformity or stenosis may lead to spinal cord compression.1
Skeletal Dysplasias
Achondroplasia
The cause of hypotonia remains unclear regarding whether it is constitutional or the result of a partial neurologic deficit. There was speculation that foramen magnum stenosis might be the cause of the hypotonia, but this has been shown not to be the case.2 Some hypotonia may result from spinal cord compression at the foramen magnum because there is a relative increased frequency of sleep apnea in infants with achondroplasia, sometimes leading to sudden death.3 Sleep apnea monitors are often used for the first several months in many of these infants, and if sleep apnea is clinically significant, evaluation of potential spinal cord compression at the foramen magnum is needed.4,5 This evaluation includes not only careful recording of sleep monitor findings, but also sleep laboratory studies for apnea and oxygen saturation levels and computed tomography (CT) and magnetic resonance imaging (MRI) of the brainstem and upper cervical spine.
Somatosensory evoked potential (SSEP) monitoring has been shown to be helpful in establishing the diagnosis of cervical myelopathy in these patients.6 If SSEP testing is done, subcortical recording of SSEPs from stimulation of the median nerve is more sensitive and specific in diagnosing high cervical myelopathy than stimulation of the posterior tibial nerve.7
Previous studies have divided this foramen magnum area compression into two major types—the first involving cervical cord compromise from direct impingement of the posterior rim of the foramen magnum and the second caused by the posterior foramen magnum rim invaginating into the ring of the atlas. In these studies, it is stressed that the neural compression is of the high cervical spinal cord, not of the brainstem itself.8 Autopsy studies have noted histologic changes in the upper cervical spinal cord similar to changes seen in the central cord syndrome, and some authors believe that if one avoids placing the head of a young child with achondroplasia in hyperextension, the risk of spinal compression is lessened.9 Persistent foramen magnum compression may play a role in the later finding of a syrinx in the cervical spinal cord.10
Some degree of foramen magnum stenosis is present in essentially all children with achondroplasia. By using CT to measure the size of the foramen magnum, it has been shown that 96% have a foramen magnum size smaller than 3 standard deviations below the mean.8 How and whom to treat remains unclear, however. One study of 32 children with achondroplasia showed 28% with a history of sleep apnea and 22% with abnormal sleep study results, both of which improved in the 6 children who had foramen magnum decompression.6 Foramen magnum decompression has been reported to be performed more safely and successfully when combined with external ventricular drainage to manage the abnormal cerebrospinal fluid dynamics in this compressive condition.11 Diverse symptoms and signs such as ataxia, incontinence, and respiratory problems have been reported to be successfully treated by foramen magnum decompression and atlas laminectomy in patients ranging in age from 7 months to 30 years.12 Some authors recommend that prophylactic cervicomedullary decompression be done, even in asymptomatic children, if T2-weighted MRI signal changes in the spinal cord are present.13
Although successful foramen magnum decompression has been reported in infants,14 other authors maintain that if appropriate sleep apnea monitoring is continued until the child is 2 or 3 years old, there is a natural relative increase in the foramen magnum size with growth, relieving enough of the spinal cord compression to avoid the need for surgical treatment.15 Reported mortality and morbidity rates from foramen magnum decompression are thought by these authors to be greater than if no treatment except sleep monitoring is done. It is rarely necessary to perform foramen magnum decompression in older children or in adults. If a child has required cervicomedullary decompression, there seems to be an increased risk for symptomatic thoracolumbar stenosis requiring laminectomies before adolescence.16
In the cervical spine below the foramen magnum in achondroplasia, the principal spinal disorder is diffuse spinal stenosis. Although a small spinal canal is present from birth, signs or symptoms of neural compression in the lower cervical spine are usually not noted until middle age or later.17 At those ages, neural compression results from osteophytes that develop with time from degenerative disc changes. The cumulative effect of previous small cervical spine dimensions and osteophyte compression leads to neurologic deficits that require treatment. If pain or sensory changes in the upper extremities are the only findings, conservative care with a cervical orthosis and anti-inflammatory medications is used initially.
If a motor deficit in the upper or lower extremities is present and pain is unrelieved by nonoperative treatment, laminectomy at multiple levels is needed.18,19 MRI defines the levels of compression, which are usually multiple. As a result, when laminectomy of the cervical spine is needed in achondroplasia, often most of the cervical spine below the axis needs to have the laminae removed to relieve the neural compression. In addition, these patients may need laminectomies in the thoracic and lumbar spine, and some patients with achondroplasia require laminectomy decompression from the skull to the sacrum.20 Foraminotomies are needed at levels shown on MRI to have neural foraminal stenosis from adjacent osteophytes. After multilevel laminectomy, cervical spine fusion is generally not needed in adults, but in the rare instance that cervical laminectomy in a child with achondroplasia should become needed, there is an increased chance of postlaminectomy kyphosis, and careful follow-up evaluation is needed. Although atlantoaxial instability is commonly seen in some other skeletal dysplasias, this instability is rarely seen in achondroplasia.21,22
Of all the spinal segments, the middle and upper thoracic spine is the least involved in spinal deformity or cord compression in achondroplasia. The most common spinal deformities and stenosis, with subsequent neurologic problems, occur in the thoracolumbar and lumbar spine. Thoracolumbar kyphosis is usually present at birth to some degree but is more noticeable when the infant begins to sit (Fig. 32–1A). As sitting begins, the entire spine appears kyphotic, at least partly owing to the generalized hypotonia present at this age. Although some authors have advocated limiting infants to a reclined, rather than a fully upright, sitting position to avoid the development of thoracolumbar kyphosis,23 this does not seem to be necessary in clinical practice. In more than 90% of children with achondroplasia, thoracolumbar kyphosis improves without treatment as the standing position is assumed and lumbar lordosis develops. Because walking typically is achieved by 18 to 24 months of age, this is the time that the resolution of the thoracolumbar kyphosis begins, followed by the gradual continual improvement over the subsequent 2 to 3 years.
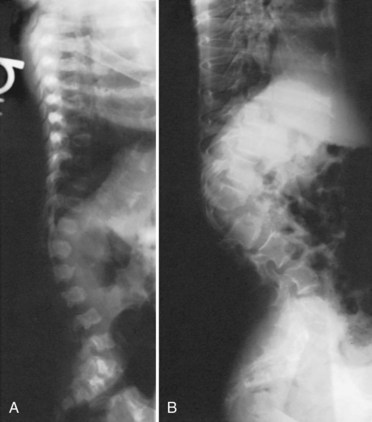
A lateral spinal radiograph shows initial relative anterior wedging of the thoracolumbar vertebrae, but this generally resolves as standing and walking occur, with the radiograph showing a gradual filling in of the anterior aspects of the vertebral bodies at the thoracolumbar apex. In the author’s experience, bracing to correct this kyphosis has limited value, and the use of a thoracolumbosacral orthosis (TLSO) is poorly tolerated by many children with achondroplasia, partly owing to the difficulty they have with TLSO wear in reaching their feet owing to their short extremities. An orthosis that uses a soft front while supporting the kyphosis has been reported to be successful,24–27 although proper patient selection for bracing remains problematic. The use of stretching exercises for the hip flexion contractures (always present in this condition), as a means to decrease lumbar lordosis and control the thoracolumbar kyphosis, has been advocated, but documentation of the effectiveness of this passive stretching program is difficult, particularly when most thoracolumbar kyphoses resolve with no treatment.28
In a few young patients with achondroplasia, thoracolumbar kyphosis does not improve and becomes progressively worse with time. If there is significant wedging of one or more of the thoracolumbar vertebrae at 5 or 6 years of age, surgical treatment is needed to allow partial correction and prevent continued progression of the deformity (Fig. 32–1B).29 This approach is based on review of early childhood x-rays of teenagers later requiring treatment for severe thoracolumbar kyphosis and neurologic deficits. It is generally possible by age 5 or 6 years to determine whether or not the thoracolumbar wedging would improve without treatment or progress to a more severe deformity. Using this approach at this earlier age, the kyphotic deformity generally can be corrected better and more safely, leading to the prevention of localized increased kyphosis and early spinal cord compression.
As thoracolumbar kyphosis increases, so does the compensatory lumbar lordosis. Because the lumbar spine capacity decreases as lumbar lordosis increases, control of the thoracolumbar kyphosis seems to delay the onset of lumbar spinal stenosis symptoms. Whether this early kyphosis fusion will eventually lead to less of a need for decompressive lumbar laminectomy has not been determined. Circumferential fusion of the kyphosis thoracolumbar area in children younger than 2 years has been reported to lead to hypoplastic vertebral bodies and iatrogenic spinal stenosis, apparently as a result of inhibition of circumferential vertebral growth after the fusion.30 In an average-sized individual, it has been shown, however, that the spinal canal achieves its adult dimensions by age 6 years, so circumferential fusion after this age should not lead to iatrogenic stenosis.
In young children with persistent thoracolumbar kyphosis at age 6, the author’s favored surgical technique involves a combined anterior and posterior spinal fusion on the same operative day.29 The child is positioned with a beanbag and tape in a lateral decubitus position with the table tilted about 20 degrees, to allow for simultaneous exposures for the anterior and posterior spinal surgery. Through a thoracoretroperitoneal approach, anterior discectomies are done at three or four levels at the apex of the kyphotic deformity. Because the surgical approach at this level commonly involves an approach through the rib bed of either the 10th or the 11th rib, the portion of the rib removed is saved for an anterior strut graft.
In the author’s surgical series of more than 25 patients with achondroplasia with kyphotic deformity requiring surgical treatment, more than half have temporarily lost SSEPs at the time of the initial correction of the kyphosis. None of these patients have had permanent neurologic deficit postoperatively. If the SSEPs are lost, however, the rod must be removed and bent into more kyphosis before reinsertion of the rods. Postoperatively, a TLSO brace may be used for about 3 months during the day to allow for some protection for the instrumentation and fusion, but the child remains ambulatory at all times. After 3 months postoperatively, sports activity restrictions are stopped, and full activity is resumed (Fig. 32–2). In another series of 12 immature patients with achondroplasia treated with pedicle screw posterior instrumentation and fusion, no loss of evoked potentials was noted, and overall mean kyphosis correction was 50%.31 The exact cause of this neurologic compromise is unclear but may be related to buckling of the ligamentum flavum with kyphosis correction, leading to further spinal canal compromise.
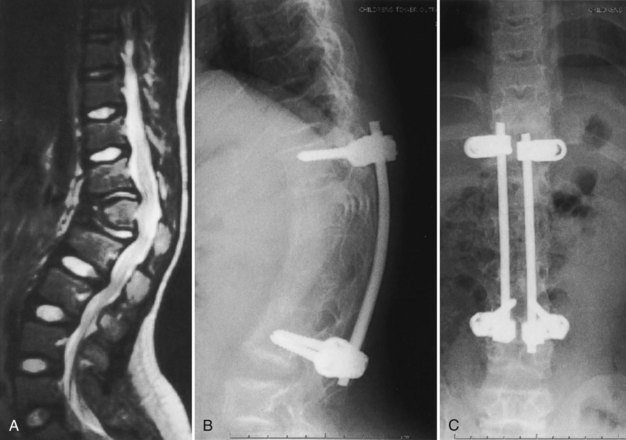
An alternative approach has been reported to treat persistent thoracolumbar kyphosis in which spinal instrumentation is placed in the anterior spine rather than in the posterior spine.32 An anterior strut bone graft is placed, and posterior fusion, without instrumentation, is done. This posterior fusion is repeated about 4 months after the initial surgery. A body jacket cast is used for 6 months, and a brace is used for another 3 months. Using this approach in four patients, Ain and Shirley32 obtained solid fusion, kyphosis correction of 23% to 31%, and no worsening of neurologic function.
Although clinically the principal thoracic and lumbar problem in preadolescent children with achondroplasia is persistent thoracolumbar kyphosis, neurologic compromise from spinal stenosis is the most common spinal disorder in teenagers and adults. Lutter and Langer33 separated these neurologic manifestations into four types: I, progressive, insidious onset; II, intermittent claudication; III, nerve root compression; and IV, acute onset of paraplegia. Types I and II are the most common. In type I, there is a slow but progressive onset of back pain, associated with lower extremity paresthesias and sensory loss.
Urologic function is often impaired, subclinically at first but later leading to incontinence.34 If urologic problems are suspected in the initial stages, voiding cystourethrogram can help to diagnose urinary control problems. If voiding cystourethrogram is abnormal, MRI of the thoracic and lumbar spine is indicated. If significant spinal stenosis is present, laminectomy decompression is needed to reverse the urologic problems. In one series of 22 pediatric patients with achondroplasia with symptoms and signs of spinal claudication and requiring laminectomy surgery, 77% had bladder incontinence. These patients requiring laminectomy at this young age had narrower lumbar interpediculate distances and greater thoracolumbar kyphosis than young patients with achondroplasia not requiring laminectomy.35
In achondroplasia with type II with neurologic manifestations, there is also a slow, progressive onset of symptoms, with the patient first noting a decreased ability to walk distances. Pain and weakness in the legs result when standing and walking occur, and these symptoms are relieved by the patient squatting, sitting, leaning forward, or lying down. These all are spinal positional changes that flex the lumbar spine to allow more room for the cauda equina and relieve the spinal claudication symptoms. It has been shown in a stillborn infant with achondroplasia that the capacity or space within the lumbar spine is nearly doubled by fully flexing the lumbar spine when compared with the extended or lordotic position.28
For any of these types, if neurologic deficits are suspected from the history or shown on physical examination, MRI is indicated to localize the site of abnormality. The problem most common in the interpretation of MRI is to determine which level is causing the neurologic signs or symptoms because there is diffuse stenosis usually present in the lower thoracic spine and the entire lumbar spine. Imaging studies show that the primary stenosis is from the narrowing of the interpediculate distances because the anteroposterior dimension of the spinal canal is relatively more normal until osteophytes from disc degeneration protrude into the spinal canal.36,37 Occasionally, CT myelography may be used to evaluate for levels of spinal stenosis, but if this is done, the myelographic dye should be placed via cisternal puncture in the upper cervical spine and not by lumbar puncture because lumbar puncture, with loss of cerebrospinal fluid, may lead to increased neurologic deficits in achondroplasia.38
The selection of the surgical procedures best suited to the individual patient with achondroplasia depends on the physical examination and the imaging findings. In a patient with type III neurologic manifestations, a limited laminectomy, disc excision, and foraminotomy generally suffice to relieve the symptoms. More commonly, in the other types of neurologic manifestations, multilevel decompressive laminectomy is the surgical treatment of choice.39,40 Laminoplasty was reported to be successful for complete relief of symptoms in 71% of 35 patients with achondroplasia and lumbar stenosis, but the use of this procedure has not been widespread.41 Even if multilevel laminectomy is done in these patients, fusion may not be needed unless there is a preexisting thoracolumbar kyphosis of about 30 degrees or more. If there is preexisting kyphosis at this level, pedicle screw fixation with dual-rod instrumentation is used with no instrumentation, no matter how small, within the spinal canal at the thoracic levels without laminectomy, although posterior pedicle screws can be used with laminectomy.
In situations with marked thoracolumbar kyphosis and multilevel stenosis on MRI, it is often difficult to determine the exact level of neurologic compromise. Generally, if depressed knee and ankle deep tendon reflexes and leg weakness are present, lumbar laminectomy seems to suffice for treatment. If there is MRI evidence of significant anterior spinal cord compression at the apex of a thoracolumbar kyphosis, and hyperreflexia is present together with leg weakness and sensory changes, anterior partial vertebrectomy or posterior pedicle subtraction osteotomy to decompress the anterior spinal cord at the apex of the kyphosis may be needed, in addition to multilevel lumbar and lower thoracic laminectomies. A report of four patients with achondroplasia and marked kyphosis who had decompression through a pedicle subtraction osteotomy noted that there was a mean kyphosis correction of 44%, but that, despite final improvement in neurologic status, transient postoperative weakness was noted in two of the four patients.42
In achondroplastic patients without coincident kyphosis but with neurologic deficits secondary to the diffuse stenosis, multilevel laminectomy is the treatment of choice (Fig. 32–3).39,40 Before surgery, it is essential to view the entire thoracic and lumbar spine on MRI to assess for all levels of stenosis that may be causing lower extremity weakness. It is important to decompress all levels that appear to have neural compression on MRI, and most often the levels for laminectomy extend from around T10 to S1. If only a single-level or double-level laminectomy is done at what appear to be the most involved areas, recurrence of new symptoms within months of the decompressive surgery is common as new levels of compression develop adjacent to the initial sites of decompression.
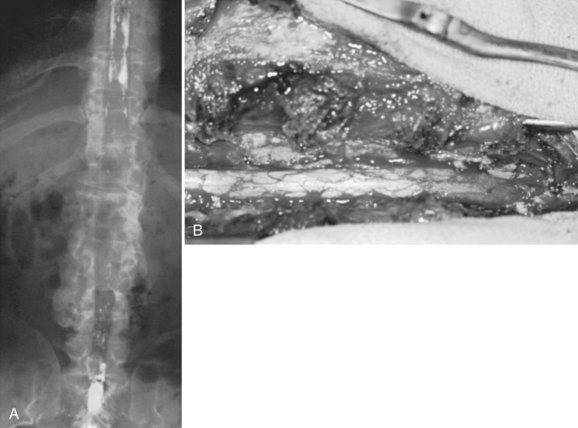
Because of the severe spinal stenosis present in achondroplasia, with the absence of epidural fat and concurrent loss of most of the subarachnoid space seen at the time of surgery, special precautions and surgical techniques are needed to complete these laminectomies more safely.34 The use of rongeurs within the spinal canal during laminectomy should be limited, owing to the severe stenosis. A postoperative increase in neurologic deficits is common, even if extreme care is taken during the laminectomy procedure. The use of a high-speed bur to transect the lamina on each side just medial to the facet or at the facet level is recommended. After the laminae have been transected, the posterior elements are lifted dorsally with a clamp, with the intent to avoid placing instruments within the stenotic spinal canal, which may injure the neural elements.
Foraminotomies can be done as needed after the laminae are removed, but MRI studies showed that although the foramina in achondroplasia lumbar spines were smaller than in control groups, the percentage of foraminal space occupied by the nerve root was similar between the two groups. The conclusion was that spinal stenosis symptoms arose from the central canal stenosis, not from foraminal stenosis. It would seem that foraminotomies in these patients are usually not needed.43 Unless there is increased kyphosis in the area of laminectomies, the author has noted that fusion may not be needed, even if laminectomies include removal of the facets. It has been reported more recently, however, that 10 skeletally immature patients with achondroplasia required spinal instrumentation and posterior fusion after laminectomies across the thoracolumbar region.44
After extensive laminectomy surgery, as described earlier, there may be areas of thinned dura that later form pseudomeningoceles and lead to later neurologic deterioration or pain or both. Using the paraspinal muscles to obliterate the dead space left by removal of the bony laminae seems to be helpful in decreasing the formation of these postlaminectomy pseudomeningoceles.34 Ain and colleagues45 reported that 61% of 98 achondroplasia patients undergoing laminectomy surgery had at least one perioperative complication, including 37% with dural tears, 23% with neurologic complications, 9% with infection, and 1 death.
Signs and symptoms of recurrent spinal stenosis may occur years after initial decompressive laminectomies. The most common cause of recurrent stenosis seems to be facet hypertrophy and disc disease, although scar tissue may form over the decompressed dural sac as well. MRI studies with gadolinium enhancement often help to visualize the scar tissue present and where the cauda equina compression has recurred. In one series of eight patients with restenosis, repeat decompression helped improve motor function in some, but three of these patients had significant complications.46
In patients with significant thoracolumbar kyphosis and lumbar spine stenosis, both of these conditions can be a possible cause of the underlying neurologic deficit.47 In this setting, MRI of the entire spine is used to ascertain all levels of compression. To treat lumbar spinal stenosis and anterior spinal cord compression at the apex of the kyphosis, anterior decompression and fusion at the apex of the kyphosis, together with a posterior multilevel laminectomy and instrumented fusion, is needed.29,47 Anterior decompression and fusion may be done through an anterior approach by vertebrectomy and strut bone graft fusion at the apex of the deformity or by an approach through the pedicles for anterior decompression with cage and bone graft stabilization. After posterior decompression at multiple levels is completed, pedicle screw and rod instrumentation is inserted, and bone graft is placed to complete the fusion. The pedicle anatomy of the thoracic and lumbar spine has been shown by CT to be markedly different from that of the normal spine. In addition to other findings reported, all pedicles are directed cranially, the pedicle starting points diverge from T9 to L5, and the maximal screw path length is at L2.48,49 Instead of stainless steel implants, titanium spinal implants are used to obtain better images at MRI if there is a subsequent need to evaluate the spinal cord further.
Diastrophic Dysplasia
Characteristic diagnostic features include micromelia with markedly short stature; “hitchhiker’s thumb”; stiff proximal interphalangeal joints of the fingers; severe equinovarus foot deformity; and, within a few weeks of birth, the formation of external ear cysts, which lead to scarring and the classic “cauliflower ear” appearance.50 Intelligence is normal. A cleft palate is present in about 25% of these children.
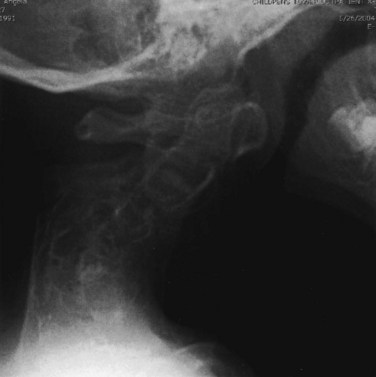
The spine has several areas of involvement in diastrophic dysplasia. All patients have spina bifida of the cervical spine, although symptoms directly related to this anatomic feature do not seem to occur.51,52 Upper cervical abnormalities seen in some of the other skeletal dysplasias, such as foramen magnum stenosis and atlantoaxial instability, are not present in diastrophic dysplasia.53 The primary cervical spine abnormality in this condition is mid-cervical kyphosis,54,55 although a review of 122 patients from Finland found only a 4% incidence of cervical kyphosis.56 At a young age, many patients with diastrophic dysplasia have mild cervical kyphosis, but most of these cases resolve with time and growth, by an average of 7 years of age.51,57
In patients with progressive cervical kyphosis, anterior and posterior fusion is recommended for optimal stabilization (Fig. 32–4). A halo brace is used to allow partial correction and postoperative immobilization until the fusion is solid. The kyphotic neck is usually relatively stiff, and aggressive attempts to correct the kyphosis are more likely to lead to iatrogenic neurologic injury. Laminectomy does not have a role here. If there is spinal cord compression that requires treatment, anterior decompression with vertebrectomies at the apex is the surgical approach of choice.
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


