CHAPTER 9 Genetic Applications
An Overview
Basics
Definition
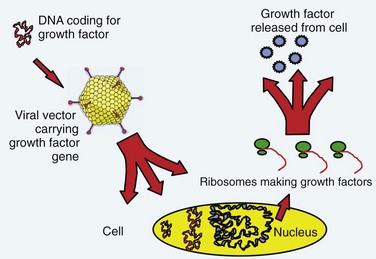
The term gene therapy was previously used to describe replacement of a defective gene with a functional copy by means of gene transfer. The diseases originally targeted for gene therapy were classic, heritable genetic disorders. The term now broadly defines therapy involving the transfer of exogenous genes (complementary DNA [cDNA]) encoding therapeutic proteins into cells to treat disease.1 The genetically altered cells are made into protein-producing “factories” churning out disease-altering gene products. Specifically, the host cell transcribes the exogenous gene (or transgene) into messenger RNA (mRNA); cytoplasmic ribosomes translate mRNA into the protein product. These products can affect not only the metabolism of cells from which they were made but also the metabolism of adjacent non–genetically altered cells via paracrine mechanisms (Fig. 9–1).
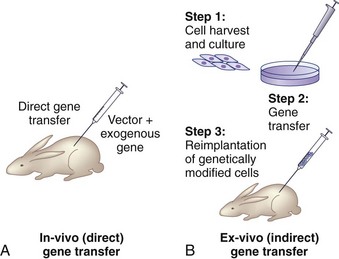
There are two basic strategies for delivering exogenous genes to target cells. The first is the in vivo method, in which a gene-carrying vector is directly transferred to an intended population of cells within the host. The second approach, known as ex vivo gene therapy, involves removing target cells from the body, genetically altering them in vitro, and reimplanting them in the body (Fig. 9–2). Ex vivo methods are more complex and involve multiple, time-intensive steps. This approach is relatively safer, however, because the genetically altered cells may be observed for abnormal behavior before implantation. The ex vivo strategy allows the opportunity for in vitro selection of cells that express the gene of interest at high levels. Gene transfer via the in vivo method is technically simpler. There are relative advantages and disadvantages to both approaches that depend on the anatomy and physiology of the target organs, the pathophysiology of the disease being treated, the vector of choice, and safety considerations.2
Nonviral Vectors
Nonviral vectors include liposomes, DNA-ligand complexes, gene guns, and microbubble-enhanced ultrasound. Liposomes are phospholipid vesicles that deliver genetic material into a cell by fusing with the cell membrane. Liposome vectors are simple, inexpensive, and safe, but drawbacks include transient expression of the transgene, cytotoxicity at higher concentrations, and low efficiency of transfection. DNA-ligand complexes and gene gun are nonpathogenic and relatively inexpensive to construct, but there is concern with lower transfer efficiencies and limited persistence of gene expression. Nishida and colleagues3 showed that ultrasound transfection with microbubbles significantly enhanced the transfection efficiency of plasmid DNA into the nucleus pulposus cells of rats in vivo, observing transgene expression up to 24 weeks. The overall transfection efficiency and level of gene expression of these nonviral vectors are generally inferior, however, to that of viral-mediated gene transfer. Consequently, most current studies involving gene therapy employ viral vectors.4
Viral Vectors
Retroviruses are small RNA viruses that replicate their genomic RNA into double-stranded DNA (dsDNA) via the action of reverse transcriptase. The dsDNA is integrated into the host genome at a random location where it is able to express transgene for the life of the cell. Exogenous dsDNA is replicated by the transduced cell and passed on to all progeny cells during cell division. Gene delivery with retroviral vectors results in stable, long-term expression because the gene is integrated into the cell’s genome. Because the integration is at a random site, however, the risk of potential mutagenesis of oncogenes exists. Until more recently, this risk was considered only a theoretical possibility, but preliminary reports from a gene therapy trial involving retroviral vectors suggest one of the enrolled subjects developed leukemia as a result of oncogene mutation.5 Another disadvantage of the most commonly used retroviral vector, the murine leukemia virus (MLV), is that it infects and transduces only actively dividing cells. For these reasons, MLV is most suited for ex vivo applications. Lentiviruses, another class of retroviruses, are capable of infecting nondividing cells. Their drawbacks lie in their wild-type pathogenicity and complex genomic configuration, which make molecular processing for gene transfer much more intricate.
Spinal Application: Fusion
Clinical Problem
Disorders of the spine often necessitate intervertebral fusion. Although internal fixation devices can successfully achieve temporary stabilization at practically all levels, long-term stability requires osseous consolidation. In contrast to fracture healing, spinal arthrodesis involves deposition of new bone in intersegmental locations that are not biologically structured for bone formation. Consequently, the nonunion rate is 40%6,7 with single-level fusions and higher when multiple levels are attempted. Although instrumentation has improved the rate of bony union, pseudarthrosis remains a considerable clinical problem. Autogenous bone graft may be scarce in volume in cases such as pediatric fusions and revision surgery. It is associated with substantial donor site morbidity. Owing to these significant obstacles to clinical success, extensive research has been directed at developing molecular therapies to facilitate intersegmental fusion.
Augmentation of Spinal Fusion with Bone Morphogenetic Proteins
Bone morphogenetic proteins (BMPs) are a group of osteoinductive cytokines that play an essential role in the formation and maturation of osseous tissues. Urist and colleagues8,9 originally recognized these proteins for their ability to form ectopic bone by inducing mesenchymal stem cell differentiation into chondrocytes and osteoblasts. Numerous subsequent animal studies have shown the ability of BMPs to enhance bone deposition at fusion sites.10–19 This was followed by several clinical trials that validated these preclinical findings. In a study by Patel and colleagues,20 patients undergoing posterolateral lumbar fusion augmented with iliac crest autograft and recombinant human BMP-7 (rhBMP-7) had better outcomes as measured by the Oswestry score and radiographic analysis than patients who received iliac crest autograft alone. In another human trial using rhBMP-2 in interbody fusion cages for single-level lumbar degenerative disc disease, patients who received cages filled with collagen sponge–delivered rhBMP-2 had superior clinical and radiographic results compared with control patients who received cages filled with autogenous bone graft.21 Various other clinical trials have similarly shown the efficacy of BMPs to enhance spinal arthrodesis.22,23
Gene Therapy for Spinal Fusion
Gene therapy represents a potential next step in the evolution of therapies directed at promoting a spinal fusion. Gene therapy techniques to deliver various BMP genes could overcome the barriers of high dosing and complex carrier systems and achieve long-term, controllable BMP expression. The transduced cells would secrete the BMP extracellularly, delivering it to the environment at physiologically appropriate doses for a sustained period, maximizing the osteoinductive potential of these growth factors. In addition, BMP expression could be regulated in a temporal fashion by using vectors with inducible promoters, which would allow the ability to control the activity of the protein tightly to the clinical setting. Another potential advantage is the capacity to deliver gene therapy for spine arthrodesis in a minimally invasive procedure with percutaneous injections to the spine. Lieberman and colleagues24 found an increase in the total volume of new bone with improved histologic quality when the gene for BMP-2 was delivered compared with rhBMP-2 protein alone.
Several studies have shown the feasibility of using gene therapy to enhance spinal fusion. Alden and colleagues25 showed new enchondral bone formation in paraspinal muscles injected with adenoviral BMP-2 constructs (Ad-BMP-2). Important observations made by this study included the absence of bone deposition distant from the injection site and the absence of neural compromise, suggesting that this approach may be safe for the clinical setting. In addition, Helm and colleagues26 documented that the direct injection of Ad-BMP-9 resulted in fusion in a rodent model without the development of nerve root compression or systemic side effects. In a rabbit model, Riew and associates27 showed that mesenchymal cells transduced with BMP-2 can promote spinal arthrodesis.
Many of these studies used first-generation adenoviral vectors for gene delivery to immunocompromised animals, allowing for sustained expression. Gene therapy experiments with immunocompetent animals have led to a relative paucity of bone formation,27 however, owing to the immune response elicited by adenoviral vectors. Further studies using second-generation adenoviral vectors or other vectors such as lentivirus could minimize these responses and maximize gene expression.
Another molecular avenue for bypassing the limitation of adenoviral immunogenicity is to deliver a gene for a factor that is “upstream” from the actions of BMP cytokines. In this way, neither efficient transduction nor sustained duration would be necessary because this factor would start a cascade of BMP activity after only a short period of expression. This intriguing strategy has been developed by Boden and colleagues,28,29 who showed that a novel intracellular transcription factor, LIM mineralization protein-1 (LMP-1), could be used to upregulate the expression of BMPs and their receptors. LMP-1 initiates a cascade of events intracellularly, which stimulates the secretion of osteoinductive factors, which increases BMP activity. All of the study animals that were implanted with peripheral blood buffy coat cells genetically modified with Ad-LMP-1 showed successful lumbar fusion.28 None of the 10 controlled rabbits had evidence of any bone formation, and the investigators concluded that local gene therapy could reliably induce spinal fusion in an immunocompetent animal.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


