13 SHOCK AND MULTIPLE ORGAN DYSFUNCTION SYNDROME
Shock is the hemodynamic manifestation of cellular metabolic insufficiency resulting from either inadequate cellular perfusion, a basic biochemical inability to properly utilize oxygen and other nutrients, or an inappropriate or amplified stimulation of cellular signaling cascades. A state of hyperinflammation, hypoperfusion, and hypermetabolism may influence the critically injured patient’s initial presentation. The common denominator in shock is a decreased utilization of oxygen by the tissues. Prolonged deficits in oxygen utilization lead to tissue injury and multiple organ dysfunction syndrome (MODS), which may progress to organ failure and death.
CATEGORIES OF SHOCK
CARDIOGENIC
Clinical Presentation.
Signs and symptoms of shock states related to cardiac pump failure include hypotension, increased systemic vascular resistance (SVR), tachycardia, decreased stroke volume index (SVI), decreased urine output (UO) and increased ventricular volumes measured by central venous pressure (CVP), right atrial pressure (RAP), and pulmonary artery occlusion pressure (PAOP). Refer to Table 13-1 for formulas and normal values of hemodynamic parameters. Assessment features include alteration in mentation, crackles (rales), increased respiratory rate, decreased bowel sounds, and pale, cool skin.
TABLE 13-1 Hemodynamic Parameters
| Parameter | Formula | Normal Values |
|---|---|---|
| CO | Heart rate × stroke volume | 4-8 L/min |
| CI | CO/Body surface area | 2.5-4 L/min/m2 |
| SVI | CI/Heart rate | 33-47 ml/beat/m2 |
| RAP | Direct measurement | 0-8 mm Hg |
| PAOP | Direct measurement | 8-12 mm Hg |
| RVEDVI |
| 60-100 ml/m2 |
| SVRI |
| 1360-2200 dyne/sec/cm−5/m2 |
| PVRI |
| <425 dyne/sec/cm−5/m2 |
| LVSWI | SVI (MAP – PAOP) × 0.0136 | 40-70 gm−m/m2/beat |
| RVSWI | SVI (PAP – RAP) × 0.0136 | 5-10 gm−m/m2/beat |

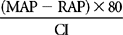
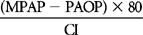
CI, Cardiac index; CO, cardiac output; LVSWI, left ventricular stroke work index; MAP, mean arterial pressure; MPAP, mean pulmonary artery pressure; PAOP, pulmonary artery occlusion pressure; PVRI, pulmonary vascular resistance index; RAP, right atrial pressure; RVEDVI, right ventricular end-diastolic volume index; RVSWI, right ventricular stroke work index; SVI, stroke volume index; SVRI, systemic vascular resistance index.
DISTRIBUTIVE
Loss of local vascular tone is also the underlying pathophysiologic mechanism of anaphylactic shock. The body reacts to a foreign substance by activating the immune system against an antigen. The inflammatory response to the antigen-antibody reaction directly alters the endothelial lining of the blood vessels, increasing vascular permeability. The antigen-antibody reaction also induces the release of histamine or histamine-like substances, which possess vasodilator properties. Histamines increase vascular capacity through venodilation, reduce arterial pressure by dilating arterioles, and increase capillary permeability, promoting a rapid shift of fluids into the interstitial spaces. Both neurogenic and anaphylactic shock can be acutely treated by infusion of intravenous fluids to restore a euvolemic state and administration of sympathomimetic drugs to promote vasoconstriction, increase venous return, and enhance myocardial contractility.
Septic shock is triggered by an overwhelming inflammatory response to infection. It is characterized by fever, elevated white blood cell count, loss of vascular endothelial integrity, hypotension despite intravascular volume administration, and an increase in oxygen requirements1 (Table 13-2). Cardiac output is elevated due to increased heart rate and vasodilation. Septic shock is discussed in detail later in this chapter.
TABLE 13-2 Definitions of Systemic Inflammatory Response Syndrome (SIRS) and Sepsis1
SIRS, Systemic inflammatory response syndrome; MODS, multiple organ dysfunction syndrome; WBC, white blood cells.
Acute adrenal insufficiency triggers distributive shock states through a lack of activation of essential catecholamines and hormones from the adrenal gland. During stress the adrenal medulla releases catecholamines such as epinephrine, norepinephrine, and dopamine, activating a sympathetic response (i.e., vasoconstriction, tachycardia). The adrenal cortex is also stimulated during stress, releasing cortisol, which promotes glyconeogenesis, and, at high levels, cortisol suppresses the inflammatory response. Aldosterone is also stimulated by the adrenal cortex; activating aldosterone increases preload. Adrenal insufficiency results in a lack of adrenal gland activity with resultant vasodilatation, hypovolemia, and lack of glucose for cellular metabolism.2
HYPOVOLEMIC SHOCK
Most often hypovolemic shock is associated with hemorrhage and a 15% to 30% blood loss.2,3 Examples of clinical conditions that may cause nonhemorrhagic hypovolemic shock include burns, intestinal disorders, and some traumatic injuries. Severe burn injury may create loss of fluids and electrolytes through denuded areas, resulting in enormous ongoing loss of plasma volume and hemoconcentration. Intestinal obstruction, ischemic bowel, and intestinal injuries may cause sequestration of fluid in the peritoneal cavity, leading to a significant decrease in circulating volume, preload, and stroke volume. The increased peritoneal volume may also cause abdominal compartment syndrome, further decreasing distal arterial blood flow and decreasing venous return. Nonhemorrhagic trauma may induce hypovolemic shock. Severe contusion or crushing damage to soft tissues often sequesters blood and plasma in the interstitial space of the injured tissues. The net effect is diminished intravascular blood volume, decreasing venous return, and stroke volume. Hypotension may not become evident until at least 30% or more of the patient blood volume is lost or the patient’s position is changed for diagnostic procedures, such as upright chest radiographs. The delay in hypotension occurs because of physiologic mechanisms that are normally able to compensate for moderate intravascular volume loss. The end result is a fall in cardiac output, intravascular volume, and oxygen delivery that are present in hypovolemic shock.
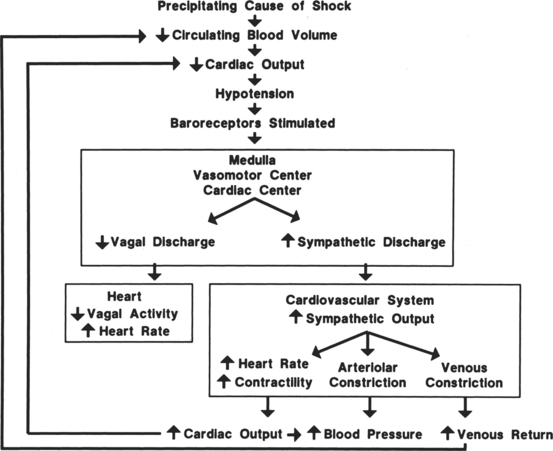
Hemorrhagic hypovolemic shock affects the patient in two ways: through a loss of circulating volume and a decrease in oxygen-carrying capacity. In addition to the loss of circulating volume, the acute decrease in red blood cells reduces both oncotic pressure and oxygen-carrying capacity. The oxygen transport deficit superimposed on the circulatory volume loss presents the characteristic features of hemorrhagic hypovolemia: significant base deficit and lactic acidosis. The initial compensatory response is a profound tachycardia combined with a compensatory systemic shunt as long as central and local sympathetic tone are intact (Figure 13-1). The vasoconstrictor response may significantly and urgently place the nonessential organs (e.g., intestines, kidney, skin) at risk of severe underperfusion.
COMMON SHOCK PATHWAYS
Regardless of the pathology, when tissue perfusion is inadequate to meet the tissue metabolic demands, a shock state can ensue. Hypoperfusion promotes a response heralded by a compensatory increase in circulating catecholamines, which augments oxygen delivery. The additional availability of oxygen supports an increase in cellular extraction of oxygen to meet higher metabolic requirements.3–5 The response to increased circulating catecholamines causes an increase in venous return and thus preload; arteriole vasoconstriction (raising afterload); increased heart rate; and enhanced myocardial contractility. Combined, these compensatory mechanisms also alter the myocardial balance of systole to diastole and increase myocardial oxygen demand and requirement for high-energy phosphate bonds.
As a result of injury, prolonged hypoxic and hypoperfused states, or sepsis, complex inflammatory pathways are stimulated. These pathways, when overactivated or unregulated, may result in catastrophic consequences. Humoral and chemical mediators change the vascular structures and metabolic pathways to such a degree that capillary blood flow and oxygen delivery mechanisms may be significantly impaired and a profound metabolic acidosis ensues. Even if oxyhemoglobin is available in the arterial bed, metabolic dysfunction, altered capillary flow, and intracellular dysfunction may prevent the extraction of oxygen, leading to organ dysfunction and eventually organ failure.5
The hypothalamic-pituitary-adrenal axis is activated during critical illness, trauma, and infection/sepsis. Effective neuroendocrine functioning is vital for a patient to adapt to the stress of illness and maintain normal cellular homeostasis. In critical illness, there is an alteration in the normal corticosteroid response, which results in the patient’s inability to increase cortisol production.2 This is also seen during sepsis as inflammatory cytokines, such as tumor necrosis factor, promote corticosteroid resistance and suppress cortisol-releasing hormone leading to adrenal insufficiency.2
COMPENSATORY RESPONSES TO HEMORRHAGE
Hemorrhage, regardless of its cause, results in a compensatory response designed to stabilize a life-threatening situation. Blood loss of less than 10% may occur without any significant effect on blood pressure or cardiac index. Blood loss of greater than 10% initiates a powerful response designed to maintain perfusion of vital organs until the circulating volume and oxygen delivery are restored either by replacement of blood and fluid during resuscitation or through a redistribution of fluid volumes in the body. When more than 10% to 15% of the total blood volume is lost, cardiac index diminishes as a result of decreased venous return. Venous return is a major determinant of preload (stretch of ventricular fibers). A proportional relationship exists between preload (ventricular filling) and stroke volume. A reduction in preload reduces stroke volume. Failure to maintain cardiac index leads to decreased arterial pressure, which causes an increase in sympathetic discharge. The sympathetic stimulation increases the heart rate and promotes vasoconstriction, which may further limit ventricular filling and ejection. Hemorrhagic loss of more than 40% of the total blood volume precipitates a profound decrease in the cardiac index and blood pressure and renders the compensatory mechanisms ineffective (see Figure 13-1).
CIRCULATORY REFLEXES RELATED TO HYPOVOLEMIA
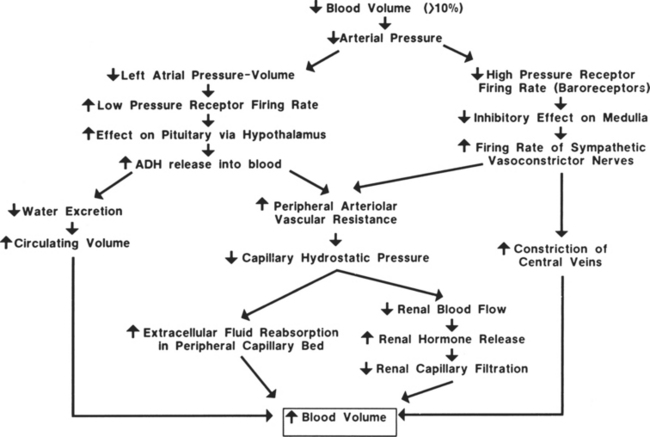
The circulatory reflexes triggered by all forms of shock, particularly acute hemorrhage, are designed to restore arterial pressure and blood flow to normal to prevent organ damage. An integrated response to intravascular volume losses greater than 10% of the total blood volume is initiated by the baroreceptors, which respond to the fall in blood pressure. Hypotension stimulates the vasomotor center, increasing sympathetic outflow. The carotid artery chemoreceptors respond to decreases in the blood pH and a rise in carbon dioxide (CO2) and the metabolic acid, H+ ion concentration. Activation of the carotid body chemoreceptors stimulates the vasomotor center and normally inhibits the autonomic nervous system (ANS) (Figure 13-2).4 The net effect of the baroreceptor response is an increased sympathetic discharge by the medullary vasomotor center, which is carried over the sympathetic efferent fibers to the heart, systemic arterioles, and adrenals. Increased sympathetic discharge has a positive inotropic effect on the myocardium that augments contractility. Improved myocardial performance results from the direct release of catecholamines at the cardiac sympathetic nerve endings in the proximity of myocardial β1-adrenergic receptors and by vasomotor center inhibition of vagal parasympathetic outflow. This change in the balance between the sympathetic and parasympathetic pathways increases atrial pacemaker activity and reduces atrioventricular conduction time, resulting in an increased heart rate. An increase in heart rate is a compensatory response mediated by sympathetic nervous system (SNS) stimulation; however, a heart rate greater than 120 beats/min may limit ventricular filling and decrease stroke volume.
Fluid Shifts
To compensate for the reduced circulating blood volume, fluids shift from extravascular to intravascular spaces. The fluid shift occurs as a direct result of the decreased intracapillary hydrostatic pressure associated with the loss of blood volume. Normally, the balance between hydrostatic pressure that promotes filtration, and the plasma oncotic pressure, which fosters reabsorption, favors a small net loss of fluid into the interstitial space. In a shock state, the reduction of blood pressure decreases the capillary hydrostatic pressure, decreasing filtration. The net effect is an increased movement of fluid volume into the capillaries. Decreased venous pressure and arteriolar vasoconstriction are separate factors contributing to the reduced capillary hydrostatic pressure and fluid movement into the vascular compartment.
Limits of Compensation
Prolonged hypovolemia (loss of more than 10% of blood volume without timely intervention) or hypovolemic shock can exhaust compensatory mechanisms so that the circulatory system cannot be maintained or restored to normal (Figure 13-3). The most important factor in patient survival is arresting the hemorrhage or cause of hypovolemia. If the hemorrhage is not stopped or volume replacement is insufficient, myocardial performance deteriorates. When the arterial pressure falls low enough, coronary blood flow decreases below that required for adequate delivery of oxygen to the myocardium. Myocardial ischemia secondary to inadequate coronary perfusion pressures reduces cardiac index, further lowering arterial pressure. The end result is deterioration of heart function.
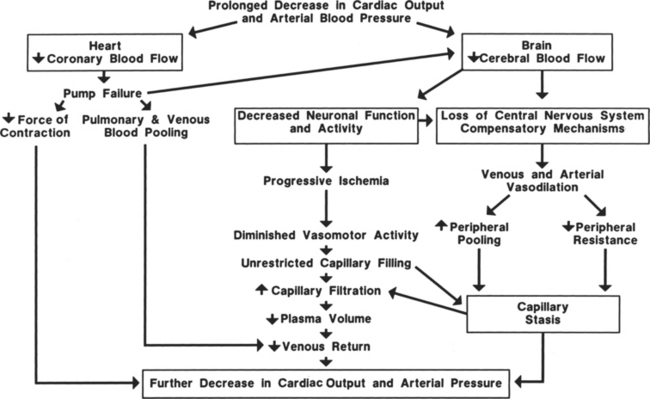
A positive-feedback cycle develops, and the initial hypovolemia leads to dire consequences. As cardiac index diminishes, cerebral perfusion is also reduced and the patient becomes increasingly obtunded. Eventually blood flow to the vasomotor center of the medulla is so compromised that the vasomotor center becomes progressively less active and finally fails. The systemic vasculature is no longer able to maintain its vascular tone; vasodilation and vascular collapse ensue. During this stage, the vascular beds behave as passive distensible tubes and no longer exhibit active adjustment to circulatory changes. Arterial vasodilation can lead to reduced blood flow to capillary beds in critical organs, whereas venous dilation causes blood to pool in the veins, decreasing venous return and further reducing cardiac output. Eventually, this sluggish blood flow in the periphery leads to changes in the capillaries that increase permeability, and large volumes of fluid move out of the vascular spaces, further lowering blood volume. Unless this cycle and these conditions are reversed rapidly, survival of the patient is doubtful. Irreversible shock occurs when damage to the cells is sufficient to depress metabolic and structural functions. At this point, widespread cellular necrosis occurs in most tissues, leading to organ failure and death.
Oxygen Consumption
Normal cell function is dependent on the delivery of adequate oxygen and other substrates to ensure adequate adenosine triphosphate (ATP) production by the mitochondria. Delivery of oxygen (DaO2) is dependent on the oxygen content in the arterial blood (CaO2) and blood flow through the tissue beds provided by the cardiac output (CO). The hemoglobin content and oxygen saturation of hemoglobin in the arterial blood (SaO2) and the partial pressure of oxygen in arterial blood (PaO2) determine CaO2. Hemoglobin and SaO2 are major determinants of CaO2 because only a very small percentage of the total oxygen content of the blood is present in a dissolved state as measured by PaO2. Table 13-3 summarizes the variables used for clinical evaluation of the adequacy of oxygen transport.
TABLE 13-3 Oxygen Delivery Parameters
| Parameter | Formula | Normal Values |
|---|---|---|
| Cao2 | (Hb × 1.37 × Sao2) + (0.003 × Pao2) | 20 ml O2/dl |
| Cvo2 | (Hb × 1.37 × Svo2) + (0.003 × Pvo2) | 15 ml O2/dl |
| Dao2I | CI × Cao2 × 10 | 500-600 ml O2/min/m2 |
| Dvo2I | CI × Cvo2 × 10 | 375-450 ml O2/min/m2 |
Cao2, Arterial oxygen content; CI, cardiac index; Cvo2, venous oxygen content; Dao2I, arterial oxygen delivery index; Dvo2I, venous oxygen delivery index; Hb, hemoglobin.
Oxygen consumption index (VO2I) is an indicator of oxygen utilization by the tissues. VO2I is affected by the adequacy of oxygen delivery and cellular ability to extract oxygen. In an afebrile, resting patient, VO2I averages 140 ml O2/min/m2 (3.5 ml O2/min/kg or 250 ml O2/min). The normal stress response to trauma, surgery, or early sepsis is associated with an increase in oxygen consumption of 15% to 35%.6–8 In a critically ill, hyperdynamic patient, cellular failures of oxidative metabolism may occur even with increased perfusion and oxygen delivery (Figure 13-4). Table 13-4 summarizes the variables used for clinical evaluation of the adequacy of oxygen utilization.
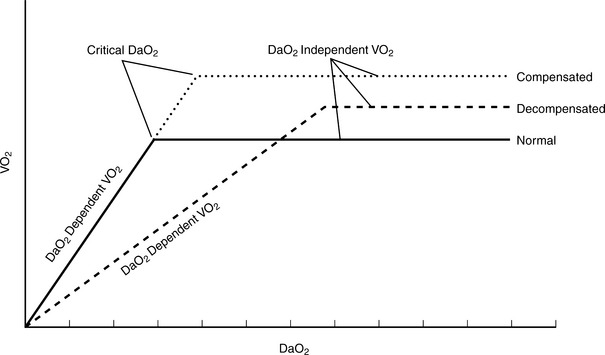
TABLE 13-4 Oxygen Utilization Parameters
| Parameter | Formula | Normal Values |
|---|---|---|
| Svo2 | Direct measurement | 60%-80% |
| Pvo2 | Direct measurement | 35-45 mm Hg |
| O2 extraction | Cao2-Cvo2 | 3-5 ml O2/dl |
| OER | Cao2-Cvo2/Cao2 | 22%-30% |
| Vo2I | (Cao2-Cvo2) × CI × 10 | 120-170 ml/min/m2 |
| pHa | Direct measurement | 7.35-7.45 |
| BE/BD | Direct measurement | −2 to −2 |
| Lactate | Direct measurement | 0.5-2.2 mmol/l |
BE/BD, Base excess/base deficit; OER, oxygen extraction ratio; pHa, arterial pH; Vo2I, oxygen consumption index; for all other abbreviations see Tables 13-1 and 13-2.
In the absence of metabolic failure, a mismatch between oxygen delivery and tissue demand is caused by reduced oxygen delivery in low perfusion syndromes.4,8 Such a condition is caused by hypovolemic or cardiogenic shock, or underperfusion of localized tissue beds as a consequence of compensatory shunting of blood flow away from less essential organs and tissues. The loss of perfusion and systemic pressure causes a reduction in oxygen delivery and thus cellular VO2 (Figure 13-5).
Oxygen availability is the most important determinant of viable oxidative energy metabolism. Cellular oxygen consumption is influenced by the availability of oxygen delivered via the microcirculation and extraction of oxygen from the capillaries to meet cellular oxygen requirements. A reduction in oxygen delivery results in several characteristic changes. A decline in blood flow results in a greater extraction of arterial oxygen in an attempt to maintain oxygen consumption. Furthermore, a compensatory increase in sympathetic stimulation results in augmented stroke volume and/or heart rate as a mechanism to increase oxygen delivery. Inadequate oxygen delivery, cellular extraction, and consumption result in cellular conversion to anaerobic metabolism for energy substrate production that causes a concomitant increase in production and release of lactate. The ability of the body to maintain a normal lactate level is an indicator of adequate resuscitation. Adversely, the higher the lactate or the longer it takes for lactate clearance, the higher the patient mortality.9–11 Therefore, lactate provides an objective, global measure to determine adequacy of tissue perfusion.
Cell Function
Maintenance of normal cellular structure and function depends on the cell’s ability to synthesize large amounts of energy. The energy requirements of different cells and organs vary depending on their functions. Continuous demands for energy are dictated by cellular and subcellular membrane ion pumps necessary to maintain the internal cellular environment; by the rate of protein synthesis, especially in cells with a high rate of protein turnover; and by mechanical work such as muscle contraction in cardiac and skeletal muscle (Figure 13-6). The energy reserves of the cell are limited, and the demand for energy is generally derived from the continual catabolism of various fuels, particularly glucose and fatty acids. The energy derived from metabolism of these fuels is stored in the form of ATP. ATP is synthesized by the phosphorylation of adenosine diphosphate (ADP) via three processes: oxidative phosphorylation using the mitochondrial electron-transport chain; substrate phosphorylation; and conversion of creatine phosphate to creatine, with the synthesis of ATP and ADP catalyzed by the enzyme creatine phosphokinase. Oxidative phosphorylation is by far the most important pathway for ATP synthesis and if interrupted by mutations or deficiencies of the proteins necessary for this process, ATP production will be severely impaired.12
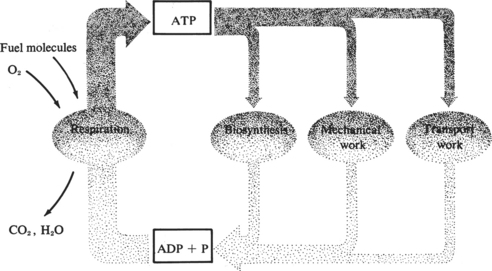
FIGURE 13-6 Relationship between energy production and energy use in maintenance of normal cell function.
(From Lehninger AL: Bio-energetics, ed 2, Menlo Park, Calif, 1971, Benjamin.)
SYSTEMIC INFLAMMATORY RESPONSE SYNDROME
PATHOPHYSIOLOGY
Persistent inflammation is fueled by the ongoing disruption of aerobic cellular cycles, which oxidize metabolic fuels to produce ATP. In the presence of SIRS, proportional use of oxygen further decreases as cellular signaling produces metabolic failure when the cells lose their ability to extract oxygen. With failure to produce these energy processes (ATP), the cells become dysfunctional and no longer maintain homeostasis. The cytokine cellular signal stimulates the immune-coagulation-inflammatory axis, which further perpetuates the hypermetabolic, hyperdynamic, and humoral response. More than 200 mediators are known to be involved with this response. Monocytes and macrophages are involved in the generation and release of the inflammatory cytokines, which are responsible for the clinical manifestations of SIRS. The major cytokines involved in SIRS include interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α), IL-6, eicosanoids, and platelet-activating factor.13 The effects of these cytokines are far-reaching and diverse. Cellular and systemic effects include alteration in vascular permeability, cellular metabolism modifications, adhesion of molecules, chemotaxis, activation of the coagulation cascade, and the production of other cytokines.14 Changes in either cellular metabolism or vascular permeability can profoundly affect the delicate balance between oxygen delivery and extraction. The decrease in vascular resistance requires a profound increase in cardiac index in order to sustain oxygen delivery and cellular oxygen uptake. Increased vascular permeability also reduces effective arterial volume and widens capillary-cell diffusion distance, further contributing to cellular ischemia. The resulting hypotension represents both the compensatory response, attempting to increase oxygen delivery to tissue, and a pathologic response, vasodilation caused by cascading mediators, procoagulation, and platelet aggregation.
CELLULAR SIGNALING, SIRS, SEPSIS, AND MODS
Historically, infection has been considered the primary cause of SIRS and MODS. However, the precipitating inflammatory response may be independent of infection. The presence of a bacterial invasion identifies a septic nidus and is termed sepsis, however; approximately 70% of patients will have clinical signs of sepsis without an apparent bacterial invader.13 In fact, any severe insult may stimulate the host-defense mechanisms implicated in the evolution of SIRS and MODS.
Activation of the body’s host-defense mechanisms precipitates a widespread inflammatory response that is not organism specific. The innate immune system provides a first-line defense against pathogens and activates a humoral-mediated response. Systemic inflammatory response syndrome results from the effects of cytokines such as TNF-α, interleukins, and from a overwhelming proinflammatory response.14 The evolution of an uncontrolled inflammatory response and the concomitant MODS is now recognized as a defect in cellular signaling.15,16
Cytokines
The first line of defense in resisting bacterial invasion is the macrophage. In addition to their phagocytic and bactericidal functions, macrophages rapidly release cytokines that activate secondary mediators of the immune system and alter the host metabolism after an inflammatory insult. Cytokines communicate messages between cells. They promote proinflammatory effects, pyogenesis and upregulation of acute- phase proteins, and an antiinflammatory response. They affect both the innate and specific (acquired) immune systems. The overlapping stimulatory and inhibitory functions among the various cytokines regulate the host’s response to injury, promoting control of the invading insult and tissue repair.17 Prolonged or exaggerated elaboration of cytokines can have deleterious effects, perpetuating SIRS and contributing to MODS. TNF, IL-1, and IL-6 have received the most attention as mediators of the inflammatory response.
TNF, primarily produced by macrophages, initiates a process that can lead to tissue injury and shock. It elicits dose-responsive hypotension, vasodilation, pyrexia, hypermetabolism, and hyperglycemia followed by hypoglycemia.14,18,19 The major effect is on the vascular endothelium, promoting adhesion of leukocytes and stimulating further cytokine secretion. In addition, TNF is a part of the acute-phase response in which there is an alteration in the plasma protein concentrations, including increased C-reactive protein lipogenesis, lipolysis, and a decrease in albumin.15,16 Chronic exposure to low plasma concentrations of TNF induces the wasting diathesis observed in various disease states. Evidence also implicates TNF in accelerating apoptosis, or programmed cell death.20–22 Although programmed cell death is a homeostatic process, its acceleration via specific cell signaling accounts for the progression from SIRS to multiple organ dysfunction.
IL-1 is an additional macrophage-derived cytokine with potent biologic effects. IL-1 is pyrogenic, accelerates the basal metabolic rate, increases oxygen consumption, augments skeletal muscle catabolism and consequently releases amino acids, reduces vascular smooth muscle reactivity, and increases the induction of IL-6 synthesis.23,24
IL-6 contributes to proinflammatory effects in the cascade; it is a potent pyrogenic, accelerates apoptosis acceleration, and impacts hepatic acute-phase protein production.23,25 IL-6 levels are significantly elevated in infected patients with SIRS and correlate with mortality in some shock states.4,26 Trauma patients appear to have initial elevation, then inhibition, of TNF and a primary elevation of IL-6.26,27 Another inflammatory cytokine, IL-8, is associated with increased lactic acid, development of disseminated intravascular coagulation (DIC), severe hypoxemia, and higher mortality in patients with severe sepsis and septic shock.24 IL-10, an anti-inflammatory cytokine, correlates with survival after severe trauma.27
Complement Activation
Bacterial infection induces a complex interaction of humoral and cellular responses. The humoral response induces a primary sequence of relatively nonspecific immunologic reactions. The early responding cytokines and the acute-phase proteins such as C-reactive protein bind to antibody proteins on the cell walls of the offending organisms. Stimulation of the complement system elicits the generation of complement split products, collectively known as anaphylatoxins. Three of the most notable are C3a, C3b, and C5a. Both C5a and C3b possess chemotactic properties for neutrophils, macrophages, and monocytes. They serve to attract these cells to areas of inflammation, enhance their adhesiveness, and promote aggregation.28 All three cell types protect against bacterial invasion by virtue of their ability to engulf bacteria via phagocytosis. Nevertheless, large-scale infiltration and activation of neutrophils, which occur with sepsis and trauma, can be detrimental to tissues rather than protective of them. Activated neutrophils release oxygen free radicals (superoxide, peroxide, and the hydroxyl radical) during phagocytosis. These toxic oxygen products normally serve a protective function by virtue of their bactericidal nature. However, if generated in sizable amounts, oxygen free radicals destroy cell membrane integrity, which ultimately leads to widespread tissue injury and impairment of organ function.
C3a induces leukocyte production of superoxides and causes extensive mast cell degranulation, which results in the liberation of histamine.28 Histamine relaxes cell-to-cell junctions, permitting the extravasation of fluid into the interstitial spaces. It is for this reason that C3a has been implicated in the genesis of permeability edema, which may contribute to organ dysfunction.4
These events facilitate bacterial ingestion by the defending white cells, which promote changes in the bacterial cell structure as well as the external environment, fostering bacterial death and lysis. Stimulation of the complement cascade has an indirect capability of triggering the activation of other enzyme cascades. These include fibrinolysis and coagulation systems, as well as the generation of prostaglandins and leukotrienes from granulocytes. These substances behave as mediators, which modulate tissue injury during sepsis and MODS. Both IL-1 and IL-2 amplify host-defense responses by stimulation of lymphocyte propagation. Moreover, these immune responses effect blast transformation and proliferation of leukocytes and promote their migration and adherence to the invading organisms and to nonviable or damaged tissues.28 Consequently the release of leukocyte proteases is increased, which completes the destruction of the bacteria and necrotic tissue, thereby facilitating their removal. Leukotrienes synthesized from white blood cells during this process also stimulate cellular immunity and alter the balance among suppressor, helper, and killer lymphocytes.29
Margination of neutrophils along vascular endothelium likely leads to damage of the cells lining the blood vessels, exposure of plasma to the underlying collagen, and activation of the coagulation cascade via Hageman factor (factor XII in the intrinsic pathway). Activated neutrophils may also cause Hageman factor formation. Hageman factor then triggers the synthesis of the vasoactive kinins. The most commonly formed is bradykinin, a potent vasodilator that also increases capillary permeability. Changes in capillary permeability and vasodilation have a role in local tissue injury and malperfusion further contributing to organ dysfunction. Activated neutrophils extrude a variety of proteases, including elastase, cathepsin G, and collagenase. These proteases degrade extracellular matrices if produced in surplus and combined with free radical production damage surrounding tissues.28
Lipid Mediators
Leukotrienes.
Collectively identified as slow-reacting substances of anaphylaxis, leukotrienes (LT) LTC4, LTD4, and LTE4 possess the ability to alter tissue permeability, produce bronchoconstriction, and trigger vasoconstriction in some vascular beds.27 The exact role of leukotrienes in sepsis remains unclear. They appear to participate in regional alterations in microvascular perfusion in addition to the exacerbation of tissue edema, particularly in the lung.30,31 Inflammation and tissue ischemia can induce generation of LTB4, a potent chemotactic agent for leukocytes. Once formed, it fosters neutrophil aggregation with release of granular constituents.31,32 Consequently, more neutrophils are attracted to the area. The aggregation of substantial numbers of cells promotes increased free radical production and perpetuates eicosanoid synthesis. Additionally, leukocyte clumping in the microvasculature can cause mechanical obstruction to blood flow.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


