Fig. 4.1
Immunolocalization of aggrecan (a), versican (b), and toluidine blue-stained proteoglycan (c) in a 14-week gestational age fetal human intervertebral disc and adjacent cartilaginous vertebral body rudiment cartilages. Higher-power images of the outer annulus fibrosus (OAF), inner annulus fibrosus (IAF), and nucleus pulposus (NP) are also presented in selected areas of the aggrecan (d–f) and versican (g–i) immunolocalizations
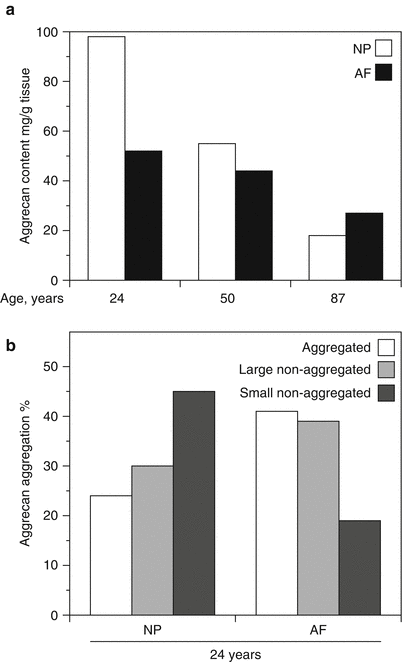
Fig. 4.2
Variation in aggrecan content (a) and aggregation (b) in the human intervertebral disc. Aggrecan content declines with age throughout the disc but at a faster rate in the nucleus pulposus than the annulus fibrosus. Aggrecan aggregation is lower and the proportion of small aggrecan fragments is greater in the nucleus pulposus (NP) than the annulus fibrosus (AF)
4.3.1 Aggrecan Protein Structure and Function
The aggrecan core protein possesses about 2,300 amino acids, which form three disulfide-bonded globular regions with two intervening extended regions (Fig. 4.3a) (Sandy et al. 1990; Watanabe et al. 1998). The amino terminal globular region (G1) responsible for interaction with HA possesses three disulfide-bonded loops. The first loop allows interaction with a link protein (LP) that stabilizes the proteoglycan aggregate (Neame and Barry 1993), and this is followed by a pair of loops responsible for the interaction with HA (Watanabe et al. 1997). The second globular region (G2) possesses two disulfide-bonded loops that share structural homology with the HA-binding loops of the G1 region. However, they do not facilitate interaction with HA (Fosang and Hardingham 1989), and their function is presently unclear. The G1 and G2 regions are separated by a short interglobular domain (IGD). After the G2 region, there is a long extended region to which the majority of GAG chains are attached. There may be over 100 GAG chains attached to each core protein, accounting for about 90 % of the molecular weight of aggrecan.
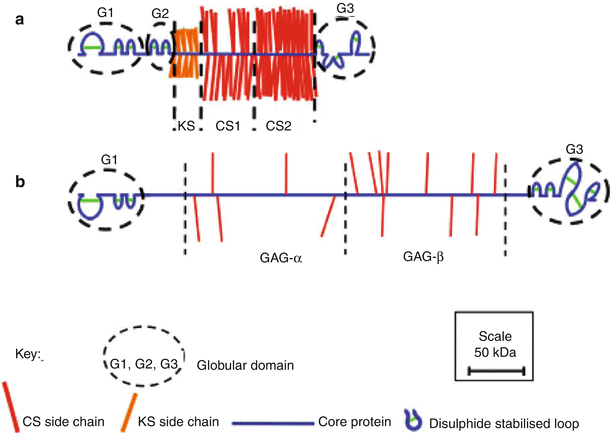
Fig. 4.3
Schematic depiction of the structural organization of aggrecan (a) and versican (b) drawn to scale
The GAG-attachment region may be divided into three domains. The domain closest to the G2 region is responsible for the attachment of KS (KS domain), and this is followed by two domains responsible for the attachment of CS (CS1 and CS2 domains). The KS and CS chains provide the aggrecan with osmotic properties essential for its function in resisting disc compression. While the CS chains are confined to the CS1 and CS2 domains, KS chains may also be present on the G1, IGD, and G2 regions (Barry et al. 1995). The CS2 domain is followed by the carboxy terminal globular domain (G3), which possesses disulfide-bonded loops having homology to epidermal growth factor (EGF), C-type lectin, and complement regulatory protein (CRP) sequences. The G3 region facilitates transit of the aggrecan through the cell during synthesis (Zheng et al. 1998) and via its lectin domain also facilitates the interaction with other components of the extracellular matrix, such as fibulins and tenascins (Day et al. 2004). It is not clear if these G3 region interactions are of functional significance in vivo, but they could potentially link proteoglycan aggregates to one another.
Both the abundance and structure of aggrecan change with age, due to variations in intracellular synthesis and extracellular degradation. Apart from possible variations in gene expression, the synthesis changes are confined to posttranslational modification of the core protein, particularly the synthesis of KS and CS (Brown et al. 1998; Roughley and White 1980). With age, the chain length of KS increases, while that of CS decreases. This could be viewed as a compensation mechanism for maintaining the sulfation of aggrecan and its swelling properties. The sulfation position of CS also changes with age, with the level of 4-sulfation decreasing and 6-sulfation increasing. It is not clear whether this variation in sulfation position has any functional significance. Currently, there is no evidence for the extracellular degradation of CS or KS, and degradative changes in aggrecan are confined to the proteolytic cleavage of its core protein (Roughley et al. 2006). Each proteolytic cleavage generates one fragment possessing a G1 region that remains bound to HA (aggregated) and one fragment that is no longer bound to HA (non-aggregated) and is free to diffuse within the disc. In articular cartilage the latter fragments are rapidly lost into the synovial fluid, but in the disc they accumulate as their diffusion is impeded by the vertebral end plates and the outer fibrous layers of the AF. With increasing age, the abundance of the non-aggregated fragments may exceed that of the aggregated fragments (Fig. 4.2b), and ongoing proteolysis further decreases the size of both the aggregated and non-aggregated fragments. Ultimately, the aggregated fragments are cleaved to their G1 region, which appears to be relatively resistant to proteolysis. As the size of the non-aggregated fragments declines, they are eventually lost from the tissue, and the total aggrecan content declines. The G1 fragments may also be eventually lost from the tissue as the size of the aggregates decreases due to depolymerization of the HA by extracellular hyaluronidases (Durigova et al. 2011b) or free radicals (Roberts et al. 1987). The average half-life of both the aggregated and non-aggregated aggrecan fragments within the disc is about 20 years (Sivan et al. 2006).
Aggrecans from different species do not possess identical structures, and there are differences in the structure of both their core proteins and GAG chains. The major core protein differences relate to the number of repeats in both the KS and CS1 domains (Barry et al. 1994; Doege et al. 1997). Of particular note is the absence of an extended KS domain in the mouse and rat, though it is unclear whether this is of functional importance. The largest species differences are in the GAG chains, which can differ enormously in chain length and degree and position of sulfation. In addition, there is variation in the abundance of aggrecan within the discs of different species. While changes in aggrecan structure and abundance are likely to have functional consequences, it is unclear whether such changes render some species more susceptible to disc or cartilage degeneration.
4.3.2 Aggrecan Gene Organization, Expression, and Mutations
The human aggrecan gene (ACAN, AGC1, CSPG1) resides on chromosome 15 (Korenberg et al. 1993) and is composed of 19 exons (Valhmu et al. 1995). Exon 1 encodes the 5′-untranslated region (UTR), exon 2 encodes the signal peptide, exons 3–6 encode the G1 region, exon 7 encodes the IGD, exons 8–10 encode the G2 region, exons 11 and 12 encode the GAG-attachment region, exons 13–18 encode the G3 region, and exon 19 encodes the 3′-UTR. The G3 region does not possess a unique structure, as the exons encoding its two EGF-like sequences and its one CRP-like sequence may undergo alternative splicing (Doege et al. 1991; Fulop et al. 1993). All alternatively spliced forms of aggrecan do, however, possess a G3 region with a lectin-like sequence, and hence may participate in ECM interactions. It is not clear whether absence of the EGF and CRP domains influences the function of the G3 domain in vivo, but this has been suggested (Day et al. 2004). Exon 12, which encodes the CS1 and CS2 domains, exhibits a unique length polymorphism within the sequence encoding the CS1 domain in the human (Doege et al. 1997). The human CS1 domain is composed of repeats of 19 amino acids, each of which bears consensus sequences for the attachment of two CS chains. The number of repeats has been reported to vary between 13 and 33, with most individuals possessing 26–28 repeats. This type of polymorphism can influence the number of CS chains present on each aggrecan molecule, and it has therefore been suggested that this may influence aggrecan function; it is predicted that those aggrecan molecules bearing less repeats are functionally inferior (Roughley 2006). This led to the prediction that individuals possessing aggrecan with a low number of CS1 repeats would be more susceptible to degeneration of both the intervertebral disc and articular cartilage. While some evidence does support this conclusion (Kawaguchi et al. 1999), it is likely that other predisposing factors must also be present.
Aggrecan gene expression is regulated by a number of factors that relate to the unique environment within the disc. TonEBP, an osmoregulatory protein present in nucleus pulposus cells, interacts with two conserved TonE motifs within the aggrecan gene promoter and promotes aggrecan synthesis, thereby allowing the nucleus pulposus cells to adapt to their hyperosmotic environment (Tsai et al. 2006). HIF-1α also enhances aggrecan promoter activity and increases nucleus pulposus cell gene expression, permitting the disc cells to function normally under low oxygen tension (Agrawal et al. 2007). In addition, both TonEBP and HIF-1α regulate the expression of the glucuronic acid transferase responsible for CS synthesis (Gogate et al. 2011; Hiyama et al. 2009). Thus, both the osmotic and hypoxic environment of the disc cells participate in maintaining normal aggrecan synthesis and structure.
Mutations in the aggrecan gene and genes involved in GAG sulfation give rise to a variety of chondrodysplasias, which affect not only the hyaline cartilages but also the intervertebral disc. In humans some forms of spondyloepiphyseal dysplasia (SED) and spondyloepimetaphyseal dysplasia (SEMD) are associated with mutations in the aggrecan gene (Gleghorn et al. 2005; Tompson et al. 2009). A nonsense mutation is responsible for nanomelia in chickens (Li et al. 1993), and a 7bp deletion in exon 5 causing a frameshift and a premature stop in exon 6 is responsible for cartilage matrix deficiency (cmd) in mice (Watanabe et al. 1994). Aggrecan is deficient in the extracellular matrix of the mutant tissues, probably due to a combination of nonsense-mediated decay of the message and impaired secretion and intracellular degradation of any truncated product. Mutations in the DSDST sulfate transporter gene are responsible for diastrophic dysplasia, atelosteogenesis type II, and achondrogenesis type 1B in humans (Karniski 2001; Superti-Furga et al. 1996), and a mutation in the APS kinase, responsible for sulfate donor (PAPS) synthesis in cartilage, gives rise to brachymorphism in mice (Kurima et al. 1998). Chondrocytes and disc cells require large amounts of sulfate for aggrecan synthesis, and when absent, an undersulfated product is formed. These disorders add credence to the view that disc function requires both a high tissue content of aggrecan and a high degree of sulfation.
4.3.3 Aggrecan Degradation in the Degenerate Disc
The interglobular domain of aggrecan is particularly susceptible to proteolysis and is cleaved by most proteinases in vitro (Fosang et al. 1992). Analysis of in vivo degradation products indicates two predominant naturally occurring cleavage sites, which could be cleaved by aggrecanases (ADAMTS4 and ADAMTS5) and matrix metalloproteinases (MMPs) (Sztrolovics et al. 1997). Both aggrecanases and several MMPs have been detected in the disc (Roberts et al. 2000), and it is currently unclear as to which family members are predominantly responsible for causing aggrecan damage in vivo. However, in vitro studies indicate that ADAMTS5 is more efficient than ADAMTS4 at cleaving within the aggrecan IGD (Gendron et al. 2007) and that MMP-3, MMP-7, and MMP-12 are the most efficient MMPs (Durigova et al. 2011a). Aggrecanases are also able to cleave within the CS2 region of aggrecan, and five cleavage sites within this region have been identified (Tortorella et al. 2002). MMPs may also cleave at sites outside the IGD, but at present the extent of their cleavage is not fully understood. One of the initial events in aggrecan degradation, following its secretion into the extracellular matrix, is removal of the G3 region (Flannery et al. 1992), and it is unclear whether aggrecanases or MMPs are responsible.
Proteolytic cleavage of aggrecan and its loss are detrimental to disc function and are thought to be directly involved with intervertebral disc degeneration (Roughley 2004). Not only does degradation and loss of aggrecan lessen the ability of the disc to swell, it may also predispose it to mechanical damage. Indeed, there may be a vicious circle in which overloading of the disc stimulates aggrecan degradation via the production of proteinases by the disc cells, which in turn renders the disc susceptible to material damage. Such material damage may be irreversible and distinguish disc degeneration from normal aging (Adams and Roughley 2006). Loss of aggrecan can also promote angiogenesis (Johnson et al. 2005) and may be a prelude to blood vessel and nerve invasion of the disc with the onset of discogenic pain (Johnson et al. 2002). Structural changes in aggrecan are also associated with the discs present in the scoliotic spine. This may also be a consequence of abnormal loading, but this time in an asymmetric manner. Indeed, aggrecan changes do vary between the concave and convex sides of the scoliotic disc. It has been suggested that dietary supplementation with CS and glucosamine may help prevent aggrecan loss in articular cartilage (Box 4.2), and if true this may also be relevant to the disc.
4.3.3.1 Box 4.2: The Therapeutic Use of Chondroitin Sulfate and Glucosamine
For the past two decades, nutraceutical companies have been promoting oral supplements of glucosamine and chondroitin sulfate (CS) for the treatment of osteoarthritis. The original theory behind this treatment was that CS was a building block for aggrecan and that glucosamine was a building block for CS and that their supplementation would therefore promote aggrecan production or their presence in the bloodstream would somehow tolerize the body to these components, thus preventing autoantibody production which is prevalent in some forms of immune-driven inflammatory arthritis. As loss of aggrecan is associated with a deterioration in the functional properties of articular cartilage in osteoarthritis, it seemed reasonable that an increase in aggrecan production would be beneficial. Indeed it may be, but the question is whether CS and glucosamine aid in this process. Aggrecan synthesis does not involve the addition of intact CS chains to its protein core, and CS synthesis does not utilize glucosamine to produce its N-acetyl galactosamine component. Commercial sources of glucosamine are derived from the chitin component (poly-N-acetyl glucosamine) of crustacean shells, and while it may make more sense to administer galactosamine as a therapeutic agent, there is no readily available commercial source of this material. Furthermore, it is likely that much of the CS entering the circulation would be degraded to its constituent monosaccharides by the liver. It is therefore not surprising that there is much skepticism concerning the ability of CS and glucosamine to promote cartilage repair, particularly given the high doses of these components required to elicit a positive response. There is however some evidence that CS and glucosamine therapy can aid in the relief of joint pain in arthritic patients, although the mechanism for this effect is not clear. If this is true, then CS and glucosamine therapy may be an attractive alternative to more conventional nonsteroidal anti-inflammatory drug (NSAID) therapy, as the former have few side effects. It does however remain to be shown whether all formulations of CS plus glucosamine are equally effective and whether CS plus glucosamine formulations are more effective than glucosamine alone.
4.4 Versican
Versican was originally identified in fibroblasts and recognized to encode a CS proteoglycan (Zimmermann and Ruoslahti 1989). Versican has a much wider tissue distribution than aggrecan and together with HA has been suggested to provide tissue hydration and viscoelasticity (Hasegawa et al. 2007). In the fetal intervertebral disc, versican is prominently expressed throughout the tissue, but not in the adjacent cartilaginous vertebral body rudiments, and it prominently demarcates the margins of the developing disc from adjacent structures (Fig. 4.1) (Smith et al. 2009). In the mature intervertebral disc, versican is present throughout the tissues, being diffusely distributed within the nucleus pulposus, and most prominent between the lamellae of the annulus fibrosus (Melrose et al. 2001). Although versican is less abundant than aggrecan in the disc, its abundance is greater than in articular cartilage (Sztrolovics et al. 2002). It is unclear whether versican serves a unique function throughout the disc, but it could provide viscoelastic properties to the outer annulus fibrosus where aggrecan is depleted. However, the function of versican may not be restricted to a structural role within the extracellular matrix, as it is also known to influence cell function, particularly in cancer (Ricciardelli et al. 2009). The versican G3 region has also been shown to influence disc cell function (Yang et al. 2003).
4.4.1 Versican Protein Structure and Function
Versican is structurally related to aggrecan, possessing terminal domains analogous to the G1 and G3 regions of aggrecan (Fig. 4.3b), although there is no evidence for alternative splicing in the versican G3 region (Grover and Roughley 1993). There is also no analogous IGD or G2 region, and the central GAG-attachment region is completely different in its amino acid sequence and GAG organization. Likewise, a domain for the attachment of KS does not exist, although KS may be present on the G1 region, and there are many fewer CS chains. The G1 region of versican has functional HA-binding and LP-binding domains, and is able to form proteoglycan aggregates, but it is uncertain whether versican and aggrecan can reside on the same aggregate. Interestingly, the four hyalectan genes reside in tandem with an LP gene (Spicer et al. 2003), suggesting that coordinated gene expression may occur. Somewhat surprisingly, versican interacts best with the LP adjacent to the aggrecan gene, whereas aggrecan interacts best with the LP adjacent to the versican gene (Shi et al. 2004). As with aggrecan, the lectin domain within the G3 region of versican has the ability to interact with fibulins and tenascins (Olin et al. 2001). The presence of multiple protein-binding motifs and the resulting possibility of versatility in function led to the name versican.
The versican core protein possesses different splice variants, which alter the size of its GAG-attachment region. The core protein of the common V1 form of versican is of a similar length to that of aggrecan, whereas that of the V0 form of versican is much larger than aggrecan, having a core protein with about 3,400 amino acids. Versican undergoes extensive proteolytic modification (Sztrolovics et al. 2002), which has hampered its purification from aggrecan. As a result, there is little information on the structure of its CS chains and whether they may change in structure with age in the disc. Although versican is commonly referred to as a CS proteoglycan, the presence of DS cannot be discounted at all ages. The spectrum of versican core protein sizes within the disc is of a similar range to those of aggrecan, varying from free G1 regions to intact molecules (Sztrolovics et al. 2002).
4.4.2 Versican Gene Organization and Mutation
The human versican gene (VCAN, CSPG2) resides on chromosome 5 (Iozzo et al. 1992) and is composed of 15 exons (Naso et al. 1994). Exon 1 encodes the 5′-UTR, exon 2 encodes the signal peptide, exons 3–6 encode the G1 region, exons 7 and 8 encode the GAG-attachment region, exons 9–14 encode the G3 region, and exon 15 encodes the 3′-UTR. The region encoded by exon 7 is referred to as GAGα and that encoded by exon 8 is referred to as GAGβ. The exons encoding the GAG-attachment region may undergo alternative splicing (Dours-Zimmermann and Zimmermann 1994), giving rise to four versican mRNAs. The presence of both the GAGα and GAGβ regions gives rise to the V0 form of versican, the presence of only the GAGβ region gives rise to the V1 form, the presence of only the GAGα region gives rise to the V2 form, and the absence of both the GAGα and GAGβ regions gives rise to the V3 form. The V1 form of versican appears to be the most common form in most tissues, including the disc (Sztrolovics et al. 2002). Whether the different forms of versican serve unique functions is unknown, but it is likely that the V3 form, being devoid of CS, may differ in function from the other forms. The V0 form of versican is analogous to PG-M, which was identified in chick limb bud mesenchyme (Ito et al. 1995).
Mutations in the human versican gene give rise to the dominantly inherited Wagner syndrome, and in the original index case, this was due to a base substitution at the exon 8/intron 8 splice junction affecting the splicing of exon 8 (Kloeckener-Gruissem et al. 2006). Additional mutations affecting the intron7/exon 8 splice junction have also been reported in other families with Wagner syndrome (Mukhopadhyay et al. 2006). Defective splicing of exon 8 results in a decrease in the V1 form of versican and an increase in the V2 and V3 forms. Wagner syndrome is classified as a vitreoretinopathy, and its ocular features are probably associated with perturbation in the role played by versican in gelling of the human vitreous. However, in accord with the widespread tissue distribution of versican, Wagner syndrome patients also exhibit extraocular features, including skeletal defects similar to those reported in Stickler syndrome. Accordingly, it is likely that perturbation in intervertebral disc formation and function will also occur.
4.4.3 Versican Degradation in the Degenerate Disc
The V1 form of versican can be cleaved by ADAMTS1 and ADAMTS4 to yield a product of 441 amino acids that includes the G1 region (Sandy et al. 2001). Thus, aggrecanases could potentially play a role in versican degradation within the disc in vivo. However, the size of this product appears to be larger than that of the free G1 region present in vivo, suggesting that other proteinases are also active. The MMPs would be the most likely candidates to fulfill this role. As with aggrecan, it is likely that premature or excessive proteolytic degradation of versican is associated with intervertebral disc degeneration. Peptide sequences within both the versican and aggrecan G1 regions have also been associated with the development of autoimmune spondyloarthropathies (Shi et al. 2003).
4.5 Perlecan
Perlecan was named for its appearance when first visualized by rotary shadowing electron microscopy, where it appeared as multiple globular domains considered to resemble a string of pearls on a chain. Perlecan has a widespread distribution throughout the developing human fetal intervertebral disc and vertebral cartilaginous rudiment cartilages, where it displays a pericellular localization pattern. However, it is also prominent in the territorial and interterritorial matrix of the developing disc (Fig. 4.4) (Smith et al. 2009). Perlecan expression is elevated in hypertrophic chondrocytes surrounding the ossification center of the developing vertebral body and in terminally differentiated growth plate chondrocytes (Smith et al. 2009). In the neonatal and adult disc, perlecan is cell associated in the pericellular matrix of the nucleus pulposus, inner and outer annulus fibrosus, cartilaginous end plates, and vertebral growth plates (Fig. 4.5). However, its relative abundance diminishes with the decline in cell number evident in the aging intervertebral disc.
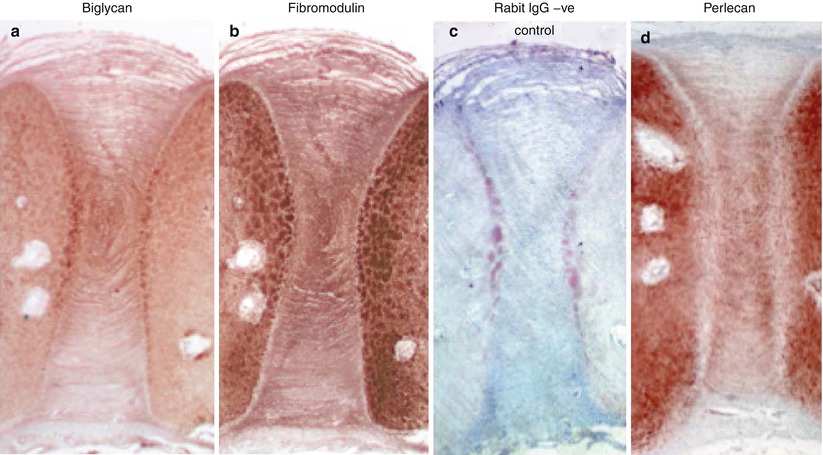
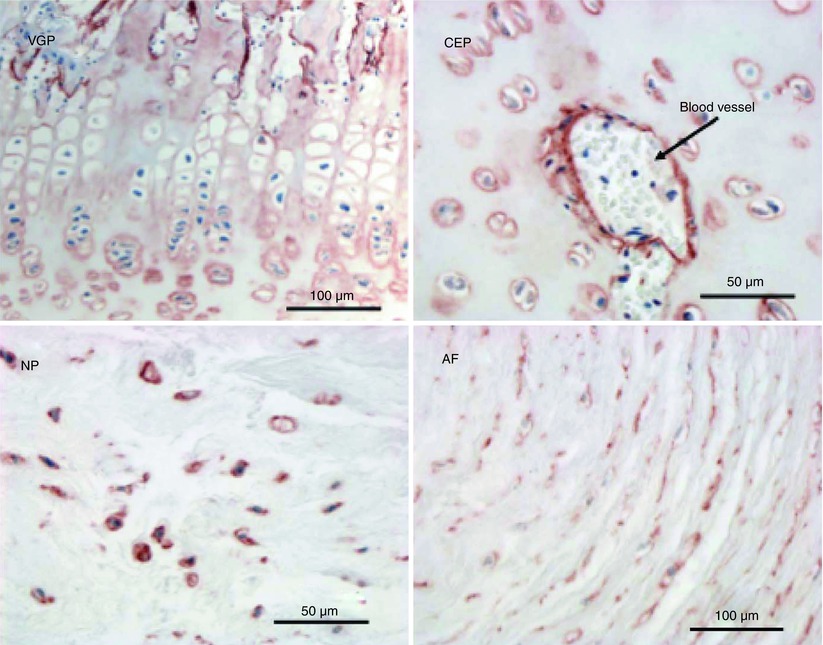
Fig. 4.4
Immunolocalization of biglycan (a), fibromodulin (b), nonimmune rabbit IgG negative control (c), and perlecan (d) in a 14-week gestational age fetal human intervertebral disc and adjacent cartilaginous vertebral body rudiment cartilages
Fig. 4.5
Immunolocalization of perlecan in the newborn ovine lumbar intervertebral disc. Perlecan is present as a pericellular proteoglycan in the vertebral growth plate (VGP), cartilaginous end plate (CEP), a blood vessel within the CEP, and the nucleus pulposus (NP) and annulus fibrosus (AF)
Perlecan interacts with a number of growth factors and morphogens, including FGF-1, FGF-2, FGF-7, FGF-9, and FGF-18; platelet-derived growth factor (PDGF); vascular endothelial cell growth factor (VEGF); hepatocyte growth factor; BMP-1, BMP-2, BMP-4, and BMP-7; hedgehog (Hh); and Wnt, and through these molecules influences cell proliferation and differentiation and matrix production (Whitelock et al. 2008). Perlecan also interacts with a number of cell attachment proteins, including laminin, fibronectin, thrombospondin, and α5β1 and α2β1 integrin, and thereby plays an important role in cell attachment and recruitment during tissue development and remodeling. Through its ability to interact with a number of matrix components, including PRELP; WARP; types IV, VI, XIII, and XVIII collagen; fibrillin-1 and fibrillin-2; nidogen-1 and nidogen-2; latent transforming growth factor-beta binding protein-2 (LTBP-2); and tropoelastin, perlecan modulates extracellular matrix assembly and stabilization (Hayes et al. 2011c; Iozzo 1994, 1998; Melrose et al. 2008b). Via these interactions, perlecan participates in disc development and in conversion of the cartilaginous vertebral rudiment cartilages into bone during spine development (Smith et al. 2009). This is consistent with roles recently ascribed to perlecan as an early chondrogenic marker in the development of cartilaginous tissues (Smith et al. 2010).
Perlecan promotes extracellular matrix production through its interactions with members of the FGF family. Perlecan is a low-affinity co-receptor for several members of the FGF family and sequesters these molecules pericellularly for later presentation to FGFRs. In this way, FGF can promote cell signaling events which drive proliferation and matrix production (Chuang et al. 2010). This sequestration process also stabilizes the FGFs, protecting them from proteolysis in situ and extending their biological half-life. Perlecan has been co-localized with FGF-18 in the developing spine, with an overexpression evident in terminally differentiated hypertrophic vertebral growth plate chondrocytes and in cells surrounding the ossification centers in the developing vertebral bodies. Perlecan associates with FGF-2 in the developing intervertebral disc interspace in the fetal human spine. Thus, FGF-18 promotes terminal differentiation of chondrocytes, leading eventually to bone formation, whereas FGF-2 maintains chondrocytes in the permanent cartilages in a delayed state of differentiation, where they are responsible for the replenishment of matrix components to effect tissue homeostasis.
Recent studies have also shown that the HS chains of perlecan are important in fibrillin and elastin assembly (Hayes et al. 2011a, c) and support earlier observations concerning basement membrane assembly. Perlecan is localized to a number of elastin-associated proteins in the intervertebral disc (Hayes et al. 2011c). LTBP-2 interacts with the perlecan HS chains (Parsi et al. 2010) and in the disc co-localizes with perlecan pericellularly. The biological significance of this localization is not known; however, LTBP-2 may have some regulatory role to play in the microfibrillogenesis process (Hirai et al. 2007; Hirani et al. 2007) by occupying sites on fibrillin-1 that LTBP-1 normally occupies (Hirani et al. 2007; Vehvilainen et al. 2009) or by interaction with another elastin-associated protein. Alternatively, by acting as a competitive substrate for the HS chains in perlecan domain I, it may regulate growth factor binding to perlecan.
4.5.1 Perlecan Protein Structure
Perlecan is a large modular HS-proteoglycan composed of five distinct domains with homology to growth factors and to protein modules involved in lipid metabolism, cell adhesion, and homotypic and heterotypic interactions involved in matrix assembly and stabilization (Melrose et al. 2008b; Murdoch and Iozzo 1993) (Box 4.3). The N-terminal domain I contains three HS attachment sites, through which HS-mediated growth factor and morphogen interactions occur. Consensus regions for GAG attachment have also been identified in the C-terminal domain V (Fig. 4.6a). The N-terminal domain is unique to perlecan, whereas domain II exhibits homology to the low-density lipoprotein receptor and domain III bears homology to the L4 laminin-type IV domain and LE laminin EGF domain. Domain IV, the largest domain in perlecan, contains multiple immunoglobulin repeats, although this domain is truncated by about 20 kDa in mouse perlecan (Melrose et al. 2008b; Murdoch and Iozzo 1993). The C-terminal domain V bears homology to the LG laminin-type G domain and contains three LG domains separated by EGF-like domains. The perlecan core protein is large (467 kDa) and highly aggregative under associative conditions, leading to the formation of higher molecular weight forms of about 800 kDa in free solution.
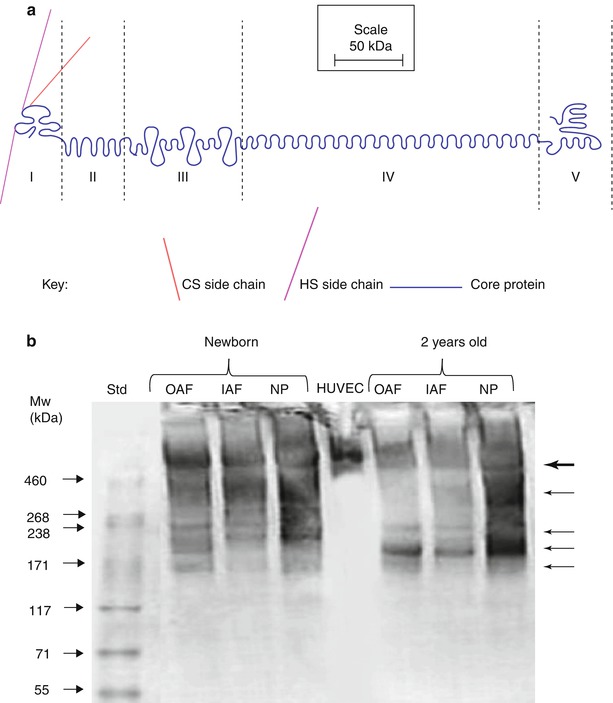
Fig. 4.6
(a) Schematic representation of the structural organization of perlecan. (b) SDS/PAGE analysis of perlecan heterogeneity in newborn and 2-year-old ovine intervertebral disc samples. Note the extensive fragmentation of disc perlecan compared to that from human vascular endothelial cells (HUVEC). The multiple perlecan species that are discernible in the disc are identified by arrows in the right hand margin. OAF outer annulus fibrosus, IAF inner annulus fibrosus, NP nucleus pulposus
The perlecan core protein can contain three HS chains in the N-terminal domain, and additional GAG consensus attachment points have been identified in domain V; however, it has yet to be definitively shown that these are occupied in the intervertebral disc. Disc cells synthesize a hybrid HS/CS proteoglycan form of perlecan, with at least one of the HS chains replaced by a chondroitin-4-sulfate (C4S) chain. In the fetal and newborn disc, this C4S chain is capped by a unique developmental CS motif identified by MAb 7D4 (Hayes et al. 2011b). However, the abundance of this 7D4 epitope on perlecan diminishes with aging, and it is not clear if this has any functional consequence.
4.5.1.1 Box 4.3: Perlecan and Its Interactive Components
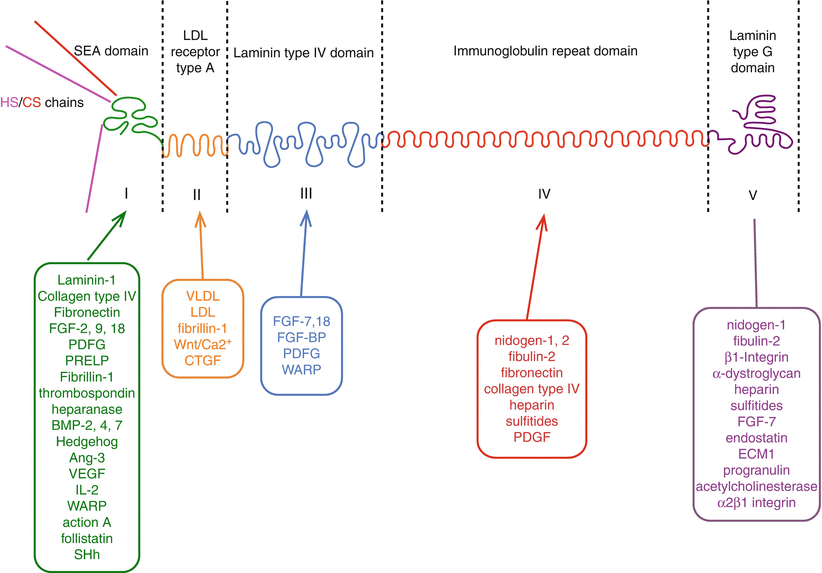
Abbreviations: FGF fibroblast growth factor, PDGF platelet derived growth factor, PRELP proline/arginine-rich and leucine-rich repeat protein/prolargin, BMP bone morphogenetic protein, Ang angiopoietin, SHh sonic hedgehog, VLDL very low density lipoprotein, LDL low density lipoprotein, Wnt a morphogenic ligand, hybrid abbreviation of Int (integration-1) and Wg (wingless), CTGF connective tissue growth factor, FGF-BP FGF binding protein, WARP von Willebrand factor A domain-related protein.
4.5.2 Perlecan Gene Organization and Mutation
The perlecan gene (HSPG2) is encoded by 94 exons located on chromosome 1p36–34 (Cohen et al. 1993; Kallunki and Tryggvason 1992; Murdoch et al. 1992; Noonan et al. 1991), with each of the structural domains being encoded by multiple exons.
The importance of perlecan in skeletogenesis (Arikawa-Hirasawa et al. 1999), vasculogenesis, and muscle and nerve development is evident from analyses of two naturally occurring mutations in the human HSPG2 gene. Schwartz-Jampel syndrome is a relatively mild skeletal condition, which arises from missense, splicing, exon skipping, and deletion mutations. These events result in partial loss of domain IV and total loss of domain V, complete loss of domain V only, or defective disulfide bonding in domain III of perlecan (Arikawa-Hirasawa et al. 2002). As a result, there are reduced functional levels of perlecan in cartilaginous tissues, chondrodysplasia, myotonia, impairment in the endochondral ossification process, and short stature. In the more severe condition of dyssegmental dysplasia, Silverman-Handmaker type, perlecan is almost undetectable in cartilaginous tissues, and this condition is characterized by a severe chondrodysplasia; severe disruption in skeletogenesis; profound effects on lung, heart, muscle, and cranial development; synaptogenesis; and complete absence of acetylcholinesterase at the neuromuscular junction leading to dystonia (Arikawa-Hirasawa et al. 2001a, b). The perlecan knockout mouse further emphasizes the essential roles of perlecan in development (Arikawa-Hirasawa et al. 1999). Perlecan knockout is a lethal condition with the majority of mouse pups dying in utero, and in those few that survive to birth, there is severe impairment in skeletal stature, cranial and long bone development, and large vessel, heart, and lung development.
Studies with the Hspg2 exon 3 null mouse (Rossi et al. 2003), where perlecan domain I containing the GAG-attachment sites is ablated, are now allowing examination of the specific role of the perlecan HS chains in skeletal development. Hspg2 exon 3 null chondrocytes are poorly responsive to FGF-2 in cell proliferation studies. Baf-32 cells transfected with FGFR3IIIc are also poorly responsive to knee rudiment cartilage perlecan predigested with heparitinase III to remove its HS chains, indicating the essential role of the domain I HS chains for FGF-2 binding and cell signaling processes (Hayes et al. 2011b). This is not the case for FGF-18, which induces cell proliferation, even in the absence of perlecan HS chains (Hayes et al. 2011b), but consistent with a domain III FGF-18 reactive site in perlecan. The Hspg2 exon 3 null mouse has a relatively mild phenotype with no apparent defects in cartilage assembly. However, recent studies have indicated that with maturation, defects become evident in cartilaginous tissues, and its reparative ability after a traumatic challenge also appears to be impaired. Fibrillin-1 assembly and deposition is also impaired in the Hspg2 exon 3 null mutant mouse intervertebral disc (Hayes et al. 2013).
4.5.3 Perlecan Degradation
Little is known about the mechanism by which perlecan is processed in the intervertebral disc. Gel electrophoresis separates full-length perlecan from three other perlecan species in newborn and young adult ovine discs. The latter three species are smaller than full-length perlecan, devoid of GAG, and detectable using a MAb to perlecan domain I. Thus, they represent fragments cleaved from the domain I carboxy terminal to the HS chains. However, the cleavage sites themselves still await detailed characterization. Perlecan fragmentation has also been observed by Western blotting of newborn and 2-year-old intervertebral disc extracts using a domain I-specific MAb, with one major and four minor species evident (Fig 4.6b). A similar range of perlecan fragments has been observed in extracts of human knee joint articular cartilage (Melrose et al. 2006).
In vitro digestion of endothelial cell perlecan with MMP-1, MMP-3, and plasmin has demonstrated the susceptibility of perlecan to cleavage in domains IV and V (Whitelock et al. 1996). Tolloid-like metalloprotease (BMP-1) also cleaves perlecan between the LG2 and LG3 domains of domain V, within the peptide sequence HLEGSGGN-↓-DAPGQYGA, releasing an anti-angiogenic peptide termed endorepellin (Bix et al. 2004, 2007; Gonzalez et al. 2005). Endorepellin has the ability to disrupt endothelial cell α2β1 integrin-based basement membrane interactions, which normally stabilize tube formation (Bix et al. 2004, 2007; Gonzalez et al. 2005). So far endorepellin is the only perlecan fragment that has been extensively characterized and its functional properties determined.
4.6 Lubricin
Lubricin was originally identified as the large mucinous glycoprotein present in synovial fluid, which by providing boundary lubrication at the surface of articular cartilage was responsible for friction-free joint motion (Swann et al. 1977). This role in joint lubrication led to the name lubricin. Later, a glycoprotein was identified in the superficial zone of articular cartilage (Schumacher et al. 1994) and termed superficial zone protein (SZP). It is produced by superficial zone chondrocytes and has been shown to be analogous in structure to lubricin (Jay et al. 2001b). Unlike lubricin, SZP has been shown to exist in part as a CS proteoglycan, termed proteoglycan 4 (PRG4). It is however not clear whether this distinction exists at all ages or in all disease states or whether the CS chain contributes to SZP function. Recently, lubricin has also been shown to reside in the intervertebral disc (Jay et al. 2001b; Shine et al. 2009; Shine and Spector 2008).
Lubricin is present in all regions of the disc but appears to be most abundant in the nucleus pulposus (Shine et al. 2009). Its core protein undergoes extensive proteolytic degradation, with accumulation of the degradation products in the tissue; it has a polydisperse size distribution comparable to other tissue sources, such as synovial fluid or articular cartilage (Fig. 4.7b). It is not known which proteinases are responsible for cleavage in vivo, but they are likely to be the same as those involved in aggrecan and versican degradation. In this respect, MMPs have been demonstrated to degrade lubricin in vitro (Elsaid et al. 2005). Also unknown is the precise function of lubricin in the disc and how proteolysis may affect lubricin function. One possibility is a role in lubricating motion between adjacent annulus fibrosus lamellae.
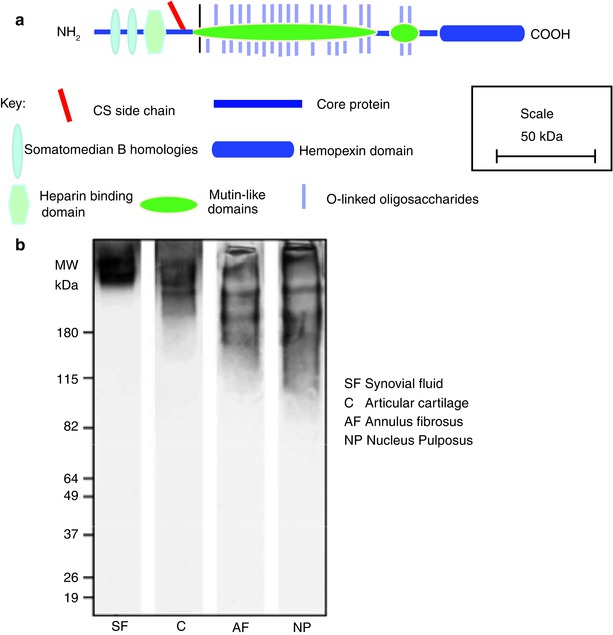
Fig. 4.7
(a) Schematic representation of the structural organization of lubricin. (b) SDS/PAGE analysis of lubricin heterogeneity in the human IVD. The lumican core protein is more fragmented in the intervertebral disc than in synovial fluid or articular cartilage
4.6.1 Lubricin Protein Structure
Intact lubricin has a molecular weight of about 240 kDa, of which about 50 % is contributed by O-linked mucin-like oligosaccharides (Swann et al. 1981b), which reside in a long central domain. This mucin-like domain is flanked by N- and C-terminal cysteine-rich domains that resemble domains of vitronectin. Domains possessing somatomedin B homology and a heparin-binding domain may reside at its N-terminus, and a hemopexin domain resides at its C-terminus (Fig. 4.7a). There is a single consensus sequence for the attachment of CS near the N-terminus of the mucin-like domain. In its SZP form, lubricin has also been reported to contain KS, but it is unclear where this resides. The lubricating properties of lubricin are conferred by its mucin-like domain (Jay et al. 2001a), but the other domains provide the potential for extracellular interactions and cell associations. The terminal domains appear to be important, as their reduction and alkylation impairs lubricin function (Swann et al. 1981a).
4.6.2 Lubricin Gene Organization and Mutation
The mRNA encoding lubricin/SZP has been shown to originate by alternative splicing of the megakaryocyte stimulating factor (MSF) precursor gene (PRG4) (Flannery et al. 1999). The human PRG4 gene resides on chromosome 1 and possesses 12 exons (Merberg et al. 1993). Exon 1 encodes the signal peptide, exons 2 and 3 encode domains with somatomedin B homology, and exons 4 and 5 encode a region containing a heparin-binding domain. Exon 6 encodes the mucin-like domain, possessing about 80 potential attachment sites for O-linked oligosaccharides and a single potential attachment site for CS. Exons 7–12 encode the carboxy terminus of the molecule, which contains a domain with hemopexin-like homology. Both synovial lubricin and cartilage SZP are derived by alternative splicing of a combination of PRG4 exons 2, 4, and 5 to yield several messages. Thus, there is no unique structure for the lubricin core protein, but all forms possess at least one somatomedin B-like domain and a hemopexin-like domain flanking the central mucin-like domain. This heterogeneity accounts for the variable size of intact lubricin.
Mutations in the human PRG4 gene give rise to the autosomal recessive camptodactyly-arthropathy-coxa vara-pericarditis syndrome (CACP) (Marcelino et al. 1999). Most of the originally identified mutations causing frameshifts or nonsense substitution near the end of exon 6 or in subsequent exons result in truncation of the hemopexin domain. These mutations result in a lack of lubricin in synovial fluid and a lack of lubricin production by cultured synoviocytes (Rhee et al. 2005b). Many of the features of CACP are recapitulated in the lubricin knockout mouse (Rhee et al. 2005a), supporting the concept that deficient production of lubricin is responsible for the CACP phenotype. The wide range of symptoms associated with CACP show that lubricin function is not restricted to joint motion but also influences tendons and the heart. While patients with CACP have spine abnormalities (Faivre et al. 2000), it is not clear whether disc function is also affected in these individuals.
4.7 The Small Leucine-Rich Repeat Proteoglycans (SLRPS)
The SLRPs are members of a large family of leucine-rich repeat (LRR) proteins, which contain multiple adjacent 24 amino acid domains bearing a common leucine-rich motif (Hocking et al. 1998). The SLRPs have been categorized into a number of subfamilies on the basis of their gene organization, number of LRRs, type of GAG substitutions, and general structural organization (Kalamajski and Oldberg 2010). Eight SLRPs have been identified in the intervertebral disc, including the CS-/DS-substituted decorin and biglycan; KS-substituted lumican, fibromodulin, and keratocan; and non-glycanated proline/arginine-rich protein (PRELP, prolargin), chondroadherin, and asporin (Fig. 4.8). These SLRPs contain ten LRRs, which are flanked by amino and carboxy terminal disulfide-bonded regions.
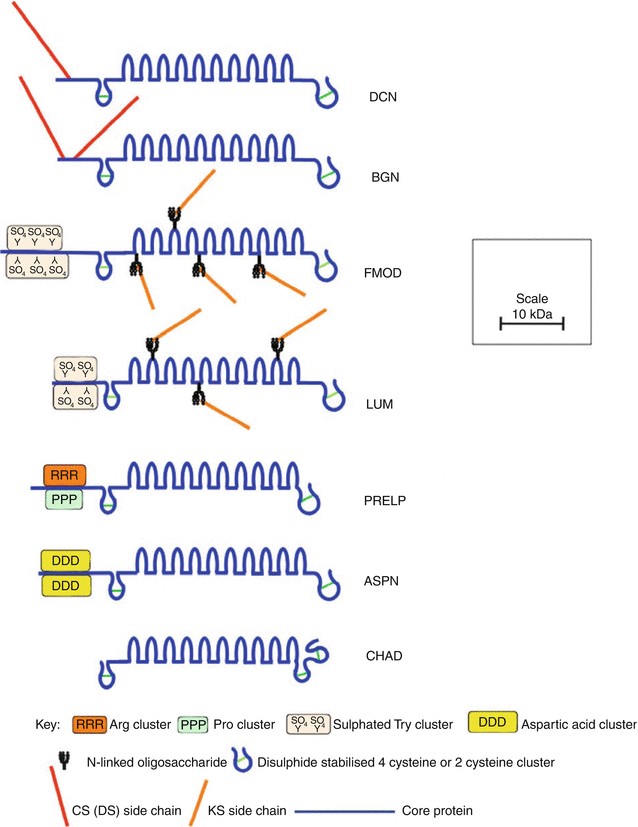
Fig. 4.8
Schematic representation of the structural organization of SLRPs found in the intervertebral disc
Decorin and biglycan possess attachment sites for their CS/DS side chains within the extreme amino terminus of their core proteins. In decorin, there is one such site, whereas biglycan contains two GAG substitution sites (Roughley and White 1989). In most connective tissues, including the intervertebral disc, the CS chains are modified in the Golgi by epimerization of the β-D-glucuronic acid moieties to α-L-iduronic acid to form DS. Non-glycanated forms of decorin and biglycan devoid of DS chains have been observed in disc tissues, and as with other connective tissues, their relative abundance accumulates with age. These non-glycanated molecules likely arise by proteolysis in the N-terminal region. Besides the removal of a small N-terminal signal peptide, the mature core proteins of decorin and biglycan are generated by removal of additional amino acid segments of 14 and 21 amino acids, respectively (Roughley et al. 1996b; Scott et al. 2000). However, the functional consequence of the removal of these pro-peptides is not known.
Lumican and fibromodulin possess four N-linked oligosaccharide chains within their central LRRs, which may be modified to KS, although substitution at all sites is uncommon (Plaas et al. 1990). Non-glycanated forms of lumican and fibromodulin also occur in connective tissues, due to lack of KS substitution (Grover et al. 1995; Roughley et al. 1996a). Only the small N-terminal signal peptides are removed from the lumican and fibromodulin core proteins to form the mature core proteins in situ. Fibromodulin and lumican also have a number of sulfated tyrosine residues clustered at their extreme N-termini in the mature protein, which may also contribute to the anionic nature of these SLRPs in situ (Antonsson et al. 1991; Onnerfjord et al. 2004; Tillgren et al. 2009).
PRELP and asporin are non-glycanated SLRPs but contain clusters of arginine and proline and aspartic acid, respectively, at their N-termini (Bengtsson et al. 1995; Grover and Roughley 1998, 2001). PRELP is unique amongst the SLRPs in possessing a cationic N-terminal region. Chondroadherin is devoid of an N-terminal core protein region where charged amino acids are clustered in PRELP and asporin. It has a similar LRR core protein structure (Grover et al. 1997), but differs from the other SLRPs in the structure of its C-terminal region.
SLRPs have important roles to play as tissue organizers based on how they affect the orientation and assembly of collagenous matrices and how they interact with growth factors and cell surface receptors during tissue development and matrix remodeling (Iozzo et al. 2011). They display diversified and overlapping functional properties with a level of redundancy between SLRP members. Asporin binds to collagen with an affinity in the low nanomolar ranges, as does decorin (Kalamajski et al. 2009). Asporin and decorin bind to the same region in fibrillar collagen and can effectively compete with one another for this site when applied at equimolar concentrations, whereas biglycan cannot (Kalamajski and Oldberg 2010). The collagen-binding sites on asporin and decorin differ, with the binding site for decorin located at LRR 7 and the binding site on asporin located at C-terminal of this site (Kalamajski et al. 2009). While lumican and fibromodulin both utilize leucine repeat domains five to seven to bind to fibrillar collagen and both can regulate early collagen fibril assembly processes, only fibromodulin facilitates growth steps leading to mature fibril formation (Kalamajski and Oldberg 2009). Decorin and biglycan display coordinated control of collagen fibrillogenesis during development and acquisition of biomechanical properties during tendon development (Iozzo et al. 2011). Alterations in the functional properties of SLRP members through point mutations affecting the structure of critical interactive regions within the LRRs can give rise to impaired tissue assembly and function.
4.7.1 Decorin
Decorin is so named since it was found to “decorate” the surface of collagen fibrils. The decorin gene (DCN) is located on chromosome 12q21.3–q23. It possesses 8 exons encoding a 36kDa core protein, which contains a single CS or DS chain located at position 4 in the mature human core protein sequence (Roughley 2006). Decorin interacts with the “d” and “e” bands of collagen I fibrils, fibronectin, C1q, EGF receptor, TGF-β, and thrombospondin. Thus, it has roles in the regulation of collagen fibrillogenesis in vitro and fibrosis in vivo and controls the bioavailability of TGF-β. Decorin also influences cell proliferation at certain stages of the cell cycle and has roles as a linking module in collagenous matrices (Iozzo and Schaefer 2010).
The regions of decorin which interact with collagen are located within the leucine-rich repeats at dissimilar regions to those involved in its interaction with TGF-β. Molecular modeling predicts that decorin possesses a “horseshoe” conformation capable of accommodating a single collagen molecule at the surface of the collagen fibrils within its concave face (Orgel et al. 2009; Scott 1996, 2003; Scott and Stockwell 2006). However, X-ray diffraction analysis of decorin crystals indicates that it exists as a dimer with interlocking concave faces (Scott et al. 2004), although it is not clear whether decorin dimers represent the functional form in solution and how this impacts its interactions with other molecules. Decorin, together with biglycan, fibromodulin, and lumican, also interacts with collagen VI, XII, and XIV (Nareyeck et al. 2004; Wiberg et al. 2002), fibronectin and elastin, and growth factors and cytokines such as EGF, IGF, TGF-β, and TNF-α. SLRP GAG chain interactions with growth factors sequester them in the matrix surrounding cells in cartilaginous matrices, with the possibility that SLRPs play a role in the regulation of the bioavailability of these growth factors.
The decorin core protein is susceptible to cleavage by MMPs in vitro (Imai et al. 1997; Monfort et al. 2006) and is fragmented in an ovine annular lesion model of intervertebral disc degeneration (Melrose et al. 2007) and in an ovine meniscectomy model of osteoarthritis in areas undergoing remodeling processes (Young et al. 2005). Fragmentation of decorin is also evident in meniscal and articular cartilages from osteoarthritic knees and hips (Melrose et al. 2008a). Decorin is processed by three isoforms of BMP-1 (von Marschall and Fisher 2010) and MT1-MMP (Mimura et al. 2009). However, by acting as a sacrificial substrate on the surface of collagen fibers or through steric exclusion effects which prevent the collagenase from accessing the collagen substrate, its presence may protect the protein from cleavage by collagenases (Geng et al. 2006).
Related posts:
 Microenvironmental Control of Disc Cell Function: Influence of Hypoxia and Osmotic Pressure
Microenvironmental Control of Disc Cell Function: Influence of Hypoxia and Osmotic Pressure
 Collagen and Other Proteins of the Nucleus Pulposus, Annulus Fibrosus, and Cartilage End Plates
Collagen and Other Proteins of the Nucleus Pulposus, Annulus Fibrosus, and Cartilage End Plates
 Intervertebral Disc Herniation: Pathophysiology and Emerging Therapies
Intervertebral Disc Herniation: Pathophysiology and Emerging Therapies
 Large Animal Models of Disc Degeneration
Large Animal Models of Disc Degeneration
 Use of Stem Cells for Regeneration of the Intervertebral Disc
Use of Stem Cells for Regeneration of the Intervertebral Disc
 Epidemiology of Lumbar Disc Degeneration
Epidemiology of Lumbar Disc Degeneration
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


