Chapter 23 Pathomechanisms of Cutaneous Lupus Erythematosus
Abnormal cutaneous reactivity to sunlight is such a seminal clinical feature of lupus erythematosus (LE) that it is one of the 11 criteria proposed by the American Rheumatism Association in 1982 for a case definition of systemic lupus erythematosus (SLE).1 Photosensitivity is also a cardinal feature of the cutaneous and neonatal forms of lupus erythematosus. This strong clinical association has led to the postulate that abnormal photoreactivity participates in the pathogenesis of cutaneous lesions in lupus erythematosus. This chapter summarizes the evidence for abnormal photoreactivity in lupus erythematosus and reviews critical and newer data on the cellular, molecular, and genetic factors that may underlie this abnormality. To enable an understanding of the potential mechanisms underlying the development of cutaneous lupus, the chapter discusses the possible interrelated roles of ultraviolet light–mediated induction of apoptosis and inflammation as well as immunomodulation. In addition, the role and importance of humoral and cellular factors in the disease process are considered. Finally, the chapter describes the participation of soluble cytokines and cofactors of inflammation in lesion induction. A model of the pathophysiology of cutaneous lupus is constructed with an incorporation of advances in the fields of photobiology, immunology, cell biology, and genetics.
Clinical Photosensitivity in Lupus
Skin lesions are common in SLE, being found in up to 90% of patients with the disease.2 Lupus-specific cutaneous findings such as malar rash (acute cutaneous lupus erythematosus [ACLE]) and discoid lupus (chronic cutaneous lupus erythematosus [CCLE]) were found in 64% and 31% of patients in a large cohort, respectively.2 Skin disease is the first symptom of disease in 23% to 28% of patients with SLE. There is a clear relationship between sunlight exposure and the manifestations of cutaneous LE, and cutaneous lesions tend to occur in sun-exposed skin. This association was first demonstrated in 1965, when Epstein used a repeated light exposure technique to show that ultraviolet (UV) radiation could induce skin lesions in patients with LE.3
Action Spectrum of Cutaneous Lupus Erythematosus
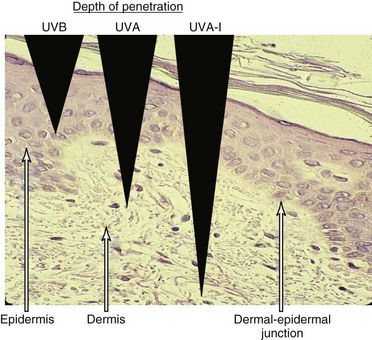
Ultraviolet light is commonly divided into germicidal UV light (UVC), midrange UV light or sunburn UV light (UVB), and long-wave UV light (UVA), also termed near-UV or black light (Figure 23-1). This separation is important because the differing wavelengths have varying biologic effects (see later). Although UVC has been used in many in vitro studies of the cellular response to UV irradiation, this spectrum of UV light is completely blocked by the earth’s atmosphere and is of dubious pathophysiologic relevance. Early investigators defined an action spectrum in the UVB range (290 to 320 nm) for the cutaneous forms of LE.4 Subsequent studies demonstrated that UVA (320 to 400 nm) also can contribute to the induction of skin lesions.5 Multicenter studies have confirmed these results.6 Although UVA-induced erythema in normal skin requires 1000 times more energy than UVB-induced erythema, daily exposure to UVA is much greater than that to UVB, and at the level of the dermal capillaries, the effect of UVA effect, as a result of greater penetrance, is much stronger than that of UVB (Figure 23-2). In formal phototesting protocols, lesions occur in a delayed fashion after UV exposure (from days to weeks) and last for weeks to months.7 In these studies, photoinducible lesions are most common in subacute cutaneous lupus erythematosus (SCLE), followed by lupus tumidus (LT) and discoid lupus erythematosus (DLE) or CCLE.6

Role of Ultraviolet Light in the Exacerbation of SLE
It is often stated that sunlight not only aggravates cutaneous LE but induces or worsens systemic features of the disease. Up to 73% of patients with SLE report photosensitivity.8 However, phototesting with standardized protocols correlates poorly with patient-reported photosensitivity,9 likely owing to the delayed nature of the lesions induced by phototesting. Repeated single patient observations indicate that sunlight may precipitate disease de novo or may aggravate existing disease. For example, use of tanning beds (a source of predominant UVA) has been reported to exacerbate SLE.10 Geographic clustering of SLE mortality has been linked to ambient solar radiation levels.11 Outdoor work, with a strongest effect among people reporting a blistering sunburn following midday sun (odds ratio [OR] = 7.9), was associated with the development of SLE in a large case-control study.12
A Selective Sensitivity to Ultraviolet Light in LE?
Clinical observations suggestive of a role for UV light in the pathogenesis of SLE and lupus skin disease have been supported by mechanistic studies. Repeated exposure to UV light can accelerate the spontaneous onset of systemic lupus in murine models. Exposure of BXSB autoimmune lupus mice to UVB has been shown to induce the release of autoantigens, to promote antibody production, and to promote early death.13 Likewise, repeated UVB exposure can induce antinuclear antibody (ANA) in autoimmune-prone NOD mice.14 UV-mediated DNA damage induces growth arrest, and DNA damage induces 45α (gadd45A) transcript expression in T cells.15 The molecule gadd45A lowers epigenetic silencing of genes by reducing methylation. The resulting hypomethylation of DNA and expression of CD11a and CD70 on T cells promotes T-cell autoreactivity and B-cell stimulation.
Responses to Ultraviolet Light in Cutaneous Lupus Erythematosus
Ultraviolet light has multiple effects on living tissue. Potential molecular targets of UV light include not only DNA but RNA, proteins, and lipids. The biologic effects of UV light on the skin are summarized in Table 23-1. In addition to alteration of DNA, cytoskeletal reorganization was noted in keratinocytes (skin cells) after UV irradiation.16 An early study by LeFeber revealed that UV light induces the binding of antibodies to selected nuclear antigens on cultured human keratinocytes.17 The specificity of these antibodies was not defined, but it is now known that they are commonly directed against Ro/SSA (anti–Sjögren syndrome antigen A), La/SSB (anti–Sjögren syndrome antigen B), ribonucleoprotein (RNP), and Smith (Sm) antigens and are the antibodies associated with SLE and photosensitivity. These results could be explained by UV-induced translocation of antigens to the cell surface with or without the death of the cell, or by other alterations in the antigens that allow the binding of autoantibodies taken up by the living cell. In 1995, Casciola-Rosen demonstrated that when keratinocytes grown in cell culture are irradiated with UVB, they actively cleave their DNA and die by a process termed apoptosis.18 During this process, the antigens recognized by autoantibodies, such as Ro/SSA, and calreticulin are concentrated in structures termed blebs or apoptotic bodies found at the cell surface. Larger blebs arise from the nucleus and harbor Ro/SSA, La/SSB, and other nuclear material. The bleb-associated antigens are then phagocytosed, packaged, and presented to dendritic cells, thereby stimulating autoimmune responses.
TABLE 23-1 Biologic Effects of Ultraviolet Radiation
| CHARACTERISTIC | ULTRAVIOLET B | ULTRAVIOLET A |
|---|---|---|
| Absorption by molecules | DNA, amino acids, melanin, urocanic acid | Melanin |
| Direct DNA damage | Increased | Minimal |
| Free radical production | Minimal | Increased |
| Depth of penetration | Epidermal | Dermal |
| Epidermal effects | Stratum corneum thickening, intermediate and delayed apoptosis, keratinocyte cytokine transcription and release | Immediate apoptosis |
| Langerhans cell effects | Inactivation, emigration | Minimal |
Ultraviolet Light, Cell Death, and the Skin
Apoptosis and necrosis are the two major mechanisms of cell death. Apoptosis is an ordered means of noninflammatory cell removal in which a central biochemical program initiates the dismantling of cells by nuclear fragmentation, formation of an apoptotic envelope, and shrinking of the cell into fragments leading to phagocytosis by parenchymal cells as well as phagocytes. In necrosis, cells are passive targets of extensive membrane damage leading to cell lysis and release of contents. UV light has long been known to induce apoptotic death in suprabasilar keratinocytes; such cells were called “sunburn cells” by morphologists.19 UV light is now known to induce such apoptosis by multiple mechanisms (for review see reference 20).
Cell Death in Cutaneous Lupus Erythematosus
Using terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick-end labeling (TUNEL) staining to detect nuclei with DNA damage, Norris demonstrated the presence of an increased number of apoptotic keratinocytes in the basal zones of CCLE lesions and in the suprabasal zones of SCLE lesions.21 The increased number of apoptotic cells could be a result of a higher rate of apoptosis induction mediated directly by UV light or as a consequence of UV-induced cytokine release. Apoptosis also can be induced by cellular cytotoxic mechanisms. Cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells can induce apoptosis through multiple mechanisms (reviewed in reference 22), including the release of perforin and granzymes, cytokine release (interferon gamma [IFN-γ], tumor necrosis factor alpha [TNF-α], TNF-β, interleukin-1 [IL-1]); and triggering of Fas by FasL. The presence of leukocytes in proximity to the apoptotic cells and of FasL-positive macrophages in proximity to apoptotic cells in lesional hair follicles suggests a role for such cellular apoptotic mechanisms in established lesions. Although detection of a higher number of apoptotic cells in LE epidermis may underlie an increase in apoptosis, either an increase in the rate of apoptotic death or a decrease in the rate of clearance of apoptotic debris could lead to the observed rise in apoptotic cell number. An accumulation of apoptotic cells in the skin of patients with CLE after UV has been associated with delayed clearance.23 Apoptotic cells are normally cleared rapidly by macrophages, and the cause of this delayed clearance in patients with CLE is still unclear. A potential role for C1q in the clearance of apoptotic debris and in the genesis of cutaneous LE is suggested by two observations. First, patients with C1q deficiency experience LE-like photosensitive eruptions.24 Second, mice with C1q deficiency demonstrate an SLE-like disease associated with an accumulation of apoptotic cells in the kidney.25
An increased number of apoptotic cells are noted in lesional LE skin. Can this finding have systemic as well as local consequences? There is evidence that the biochemical processes of apoptosis generate novel antigens that are uniquely targeted by autoantibodies. Casciola-Rosen has shown that the caspases activated during apoptosis cleave intracellular proteins into fragments that are bound by autoantibodies from some patients with LE.26 Patients with LE skin disease have more autoantibodies that preferentially recognize apoptotic-modified U1-70-kd RNP antigen than patients without skin disease.27 This finding provides further in vivo evidence that immune recognition of modified forms of self-antigen occur in cutaneous LE and suggests that this immune recognition and the processing of apoptosis-derived antigens may participate in the pathogenesis of the disease. The appearance of autoantibodies to skin-specific antigens such as desmoglein 4 in patients with “pre-SLE” suggests that the skin is an early site in the breakdown of tolerance in SLE.28
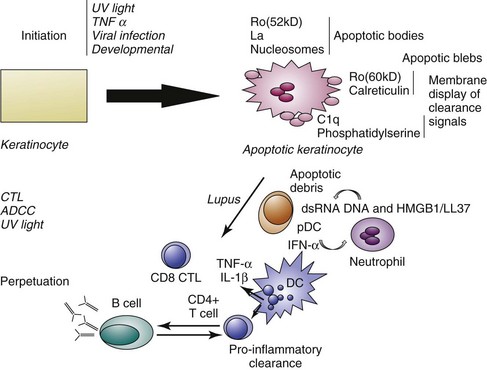
Necrosis is a cell death process characterized by the rapid depletion of adenosine triphosphate (ATP) stores and subsequent loss of cell membrane integrity that can also result from UV light injury. For example, high doses of UVB preferentially induce keratinocyte necrosis.29 Necrotic cells release potent proinflammatory mediators such as high mobility group box 1 (HMGB 1) protein30 and uric acid.31 Another form of cell death that may release antigenic material is a unique form that occurs principally in neutrophils but also in mast cells and has been termed “NETosis.” In this form of cell death, chromatin and cytoplasmic granules are released during the formation of bacteriocidal neutrophil extracellular traps (NETs). The NETs contain not only DNA but also the antimicrobial peptide LL37, which enables self DNA and RNA to engage Toll-like receptors TLR9 and TLR7 to activate plasmacytoid dendritic cells.32,33 Thus, these substances promote type 1 IFN release and autoimmunity (reviewed in reference 34). NETs are abundant in the dermis of lesional lupus skin.35 In a mouse model of lupuslike skin inflammation, neutrophil depletion results in a decrease in cytokine release following skin injury.36 An abundance of apoptotic cells and possibly necrotic cells, either from excessive amount of death induction by UV light or from a defect in clearance, could permit tolerance to self-antigens to be broken. Cells of the early inflammatory response such as neutrophils could then amplify the immune response to self-antigens. The potential role of apoptotic mechanisms in the initiation and perpetuation of photosensitive LE is summarized in Figure 23-3.
Ultraviolet Light as Inflammatory Stimulus
Erythema (redness), a normal response to UV light, is mediated by multiple eicosanoids, vasoactive mediators, neuropeptides, and cytokines released from keratinocytes, mast cells, endothelial cells, and fibroblasts. (The wide range of mediators released by UV light in the skin is listed in Table 23-2.) UV light is not only an executioner, killing keratinocytes by apoptosis/necrosis, but it also is a generator of neoantigens (such as UV-DNA) and inflammation.
TABLE 23-2 Mediator Release by Ultraviolet Radiation*
| SOURCE OF MEDIATOR | ULTRAVIOLET B | ULTRAVIOLET A |
|---|---|---|
| Keratinocyte | IL-1α, tumor necrosis factor alpha (TNF-α) | IL-8IL-10, IL-12PGE2, PGF2α |
| GM-CSF, IL-6, IL-8 | ||
| IL-10 | ||
| Transforming growth factor beta | ||
| PGE2, PGF2α | ||
| Mast cell | TNF-α | |
| LTC4, LTD4, PGD | ||
| Histamine | ||
| Endothelial cell | TNF-α, PCI2 | PCI2 |
| Langerhans cell | IL-12 |
* Ultraviolet radiation results in the release of interleukins (ILs), prostaglandins (PGs), prostacyclin (PCs), leukotrienes (LTs), and other mediators.
UV light can induce cutaneous inflammation by promoting the release of inflammatory mediators and cytokines, by inducing adhesion molecule display, and by releasing chemokines to attract inflammatory cells into the skin (reviewed in reference 37). Both UVB and UVA can participate in lesion induction and act by differing mechanisms. UVB induces the release of the primary cytokines IL-1α and TNF-α from the epidermis, initiating a cascade of inflammatory events. IL-1α and TNF-α are “primary cytokines” that induce the release of a number of other proinflammatory cytokines from the epidermis. For example, IL-1α and TNF-α induce the secondary release of IL-6, prostaglandin E2, IL-8, and granulocyte-monocyte colony-stimulating factor (GM-CSF) by keratinocytes. Chemokines are chemoattractive proteins that are associated with inflammatory cell recruitment. UVB irradiation of primary human keratinocytes in the presence of proinflammatory cytokines such as IL-1 and TNF-α significantly enhances the expression of the inflammatory chemokines CCL5, CCL20, CCL22, CCL27, and CXCL8.38
UVA upregulates IL-8 and IL-10 production in keratinocytes and FasL expression in dermal mononuclear cells. The longer wavelength of UVA allows it to penetrate into the dermis and to upregulate vascular endothelial intracellular adhesion molecule 1 (ICAM-1) and E selectin, thereby increasing leukocyte-vascular adhesion. Acute administration of low-dose UVA, but not UVB, results in IL-12 production by keratinocytes in vivo. UVA also results in a rapid increase in IFN-γ levels in the skin, the source of which may be resident epidermal T cells.39
Related posts:
 The Innate Immune System in SLE
Clinical Aspects of the Antiphospholipid Syndrome
Gastrointestinal and Hepatic Manifestations
Novel Therapies for SLE: Biological Agents Available in Practice Today
Mixed Connective Tissue Disease and Undifferentiated Connective Tissue Disease
Pathogenetic Mechanisms in Lupus Nephritis
The Innate Immune System in SLE
Clinical Aspects of the Antiphospholipid Syndrome
Gastrointestinal and Hepatic Manifestations
Novel Therapies for SLE: Biological Agents Available in Practice Today
Mixed Connective Tissue Disease and Undifferentiated Connective Tissue Disease
Pathogenetic Mechanisms in Lupus Nephritis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


