Chapter 18 Pathogenetic Mechanisms in Lupus Nephritis
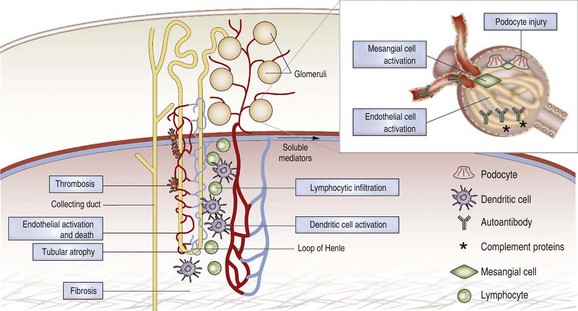
SLE nephritis is characterized by glomerular and tubulointerstitial inflammation most often initiated by the renal deposition of immune complexes1–4 that trigger a cascade of inflammatory events including complement activation,5 engagement of activating Fc receptors on mononuclear cells,6 activation of intrinsic renal cells, and recruitment of inflammatory cells. Renal deposition of autoantibodies and complement is not, however, sufficient to cause renal damage. Signals from the innate immune system and cellular immunity also contribute to renal disease. Immune complexes directly activate resident renal cells through Toll-like receptors (TLRs) to produce inflammatory mediators.7 Chemokines produced by intrinsic renal cells attract multiple subsets of inflammatory cells.8 Cytokines induce endothelial cells to express adhesion molecules, increasing the probability that they will recruit inflammatory cells after contact with immune complexes. In one animal model of nephritis, T cell–mediated interstitial renal disease and vasculitis together with mild glomerular changes occur even in the complete absence of circulating immunoglobulins.9 Similarly, in human SLE some pauci-immune forms of nephritis have been observed. Microvascular damage and thromboses also occur in the setting of SLE nephritis and may be more common in patients with antiphospholipid antibodies.10 Inadequately treated disease or repeated disease flares can lead to chronic changes, including glomerulosclerosis, tubular atrophy, and tissue fibrosis, that may ultimately lead to the death of the organ.
Renal Anatomy and Physiology
Glomerular Structure and Function
The glomerulus is an intricately organized structure that is highly permeable to water while at the same time acting as a filtration barrier for larger molecules (Figure 18-1).10a Each glomerulus consists of a spherical tuft of capillaries under arteriolar pressure that is held together by the mesangium, a supporting tissue consisting of mesangial cells and their matrix. The highly negatively charged glomerular filtration barrier consists of endothelial cells, the glomerular basement membrane, and a series of foot processes that emanate from podocytes, specialized epithelial cells located within the urinary space. Maintenance of the structural integrity of the glomerular capillary loops and of the glomerular filtration barrier depends on intricate “cross talk” between podocytes, endothelial cells, and mesangial cells; injury of any of the three cell types may lead to proteinuria. The urinary space of each glomerulus is located between and outside the podocytes and is surrounded by a single layer of parietal epithelial cells that is continuous with the podocyte layer. Importantly, the effluent blood flow from the glomerulus provides the sole blood supply for peritubular capillaries, so any decrease in glomerular blood flow will threaten the viability of the tubules.
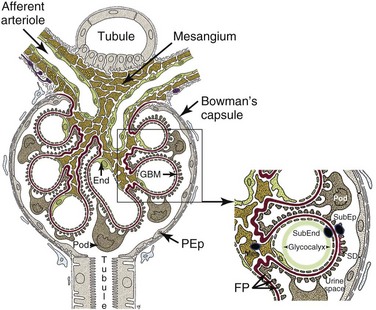
Glomerular Filtration Barrier
The glomerular filtration barrier has several layers.11 The first is a glycocalyx made up of proteoglycans and an adsorbed layer of plasma proteins that is located between the endothelial cells and the capillary lumen. Fenestrated endothelial cells form the next layer. Next is the thick glomerular basement membrane (GBM), which is synthesized by podocytes and endothelial cells and has an inner layer composed of collagen type IV and laminin sandwiched between layers of heparin sulfate. Podocyte foot processes line the epithelial side of the GBM; the intercellular junctions between adjacent foot processes are closed by the slit diaphragm, a specialized intercellular junction that acts as a molecular sieve and the final component of the filtration barrier. The slit diaphragm comprises several proteins, including nephrin, CD-associated protein (CD2AP), podocin, the tight junction protein ZO-1 (zonula occludens 1), P-cadherin, catenins, and the calcium channel TRPC6 (transient receptor potential cation channel, subfamily C, member 6), each of which is required for slit diaphragm integrity. Slit diaphragm proteins are supported by the highly dynamic podocyte actin cytoskeleton that in turn is anchored to an integrin complex that fastens each podocyte foot process to the GBM.
Podocytes
Podocytes play a crucial role in maintaining glomerular integrity and function. Apart from synthesizing the GBM, podocytes secret angiopoietin 1 and vascular endothelial growth factor 1 (VEGF-1), which help maintain the endothelial cells; loss of podocyte-derived VEGF results in renal thrombotic microangiopathy. Podocytes also express the immunoglobulin (Ig) receptor FcRn (neonatal Fc receptor), which helps clear trapped immunoglobulin, and express angiotensin receptors and angiotensin-converting enzyme (ACE).12 Inhibition of the latter pathway can improve proteinuria via systemic hemodynamic and local metabolic effects. During inflammatory states, including SLE nephritis, podocytes can be induced by TLR engagement to express B7-1, which binds to the integrin complex and displaces the foot process from the GBM, thus inducing proteinuria.13 Loss of podocytes into the urine is a feature of active SLE nephritis. Although some forms of podocyte injury are reversible, chronic injury leading to excessive podocyte loss eventually results in glomerulosclerosis and loss of renal function.13
Mesangium
The mesangium consists of mesangial cells and the mesangial matrix that contains collagens, laminin, proteoglycans, heparin sulfate, fibronectin, entactin, and nidogen. Mesangial cells are smooth muscle–like cells that contain actin and myosin; they connect to each other via gap junctions and to the GBM via cell processes. Contraction of mesangial cells regulates the size of the capillary lumen and thus the amount of glomerular blood flow. Mesangial cells are in direct contact with the vascular system via the fenestrations in the endothelial capillary cells, and their survival depends on platelet-derived growth factor (PDGF) secreted by endothelial cells. Mesangial cells produce growth factors, cytokines, adhesion molecules, chemokines, and vasoactive factors, and they express Fc receptors, C-type lectins, and some TLRs including TLR3.14,15 Mesangial deposition of immune complexes occurs in most forms of SLE nephritis, and both mesangial cell proliferation and mesangiolysis (loss of matrix) have been observed. Nevertheless, deposits restricted to the mesangium are usually associated with mild disease.
The Renal Tubules and the Kidney Interstitium
Renal Tubular Epithelial Cells
Proximal tubular epithelial cells can assume a proinflammatory and profibrotic role during chronic renal disease in which they express inflammatory mediators including complement proteins, tumor necrosis factor alpha (TNF-α), chemokines, and growth factors. Sensitive markers of renal tubular injury include kidney injury molecule 1 (KIM-1) and lipocalin 2 (LCN-2), which are both produced by proximal tubular cells and appear in the urine promptly after ischemic damage.16
Epithelial-to-mesenchymal transition is a process by which epithelial cells transform into mesenchymal cells that contribute to chronic renal fibrosis. This transition is posited to occur in response to cell injury and exposure to transforming growth factor–beta (TGF-β) and is followed by secretion of collagen and extracellular matrix. Definitive cell-tracing experiments have now shown that despite the acquisition of some mesenchymal markers, renal tubular cells do not transform into cells that cause fibrosis, calling into question the existence of this process.17
Renal Interstitial Fibroblasts
Renal fibroblasts form a structural network that helps maintain kidney architecture; they are also the source of erythropoietin. These cells form focal contacts with each other as well as capillaries and tubules. The current consensus is that renal interstitial fibroblasts are the origin of myofibroblasts that cause renal fibrosis during chronic injury.16,17 Fibroblast proliferation is enhanced by basic fibroblast growth factor (FGF-2), which is induced by TGF-β1.18
Resident Renal Mononuclear Phagocytes
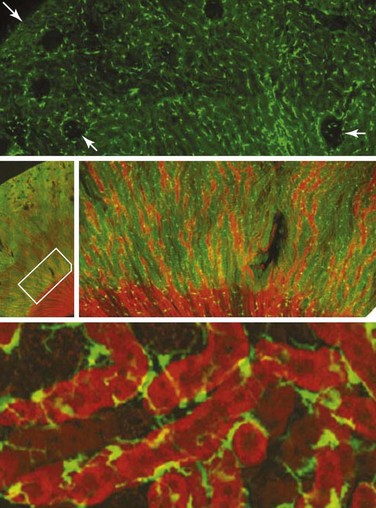
Resident renal mononuclear phagocytes have variably been called resident macrophages and resident renal dendritic cells.19 In mice, the major population of phagocytes express F4/80, CD11b, and intermediate levels of CD11c, and they are positive for major histocompatibility complex (MHC) class II molecules but express low levels of co-stimulatory molecules. The generation of CX3CR1-GFP–labeled mice has allowed visualization of these cells in a network surrounding glomerular tubules (Figure 18-2).20 These cells are capable of phagocytosis and constantly retract and extend dendritic processes into the interstitium.20,21 Their role is probably a sentinel one under physiologic circumstances, but they can contribute to renal injury once activated. In addition to these cells, other small populations of renal mononuclear phagocytes can be detected. Lymphocytes are rare in normal kidneys, with the exception of a small population of CD4+ T cells whose function is not known.
Mechanisms for Immune Complex Deposition in the Kidneys
Site of Immune Complex Deposition in SLE
Immune deposits may deposit on either side of the GBM (Figure 18-1), and both the amount and location of deposits correlate with the severity of the disease. Deposits limited to the mesangium and sparing the capillary loops are associated with International Society of Nephrology (ISN) class I and class II disease. Subendothelial deposits, located between the endothelium and the GBM, are found in ISN class III and class IV disease. These have access to the vascular space and can therefore mediate recruitment of inflammatory cells and subsequent endothelial damage. Subepithelial deposits located at the base of the podocyte foot processes outside the GBM are found in class V disease. In this type of disease, complement-mediated injury of podocytes induces them to lay down excessive matrix material that alters the structure of the basement membrane, leading to proteinuria. Inflammatory mediators recruited by subepithelial deposits are diluted into the urinary space and are excreted, limiting recruitment of inflammatory cells and local damage. Mesangial deposits are found in most classes of lupus nephritis and can induce mesangial cells to overproduce inflammatory mediators, growth factors, and extracellular matrix. Deposits of immune complexes have also been reported in the tubulointerstitium and in the vasculature.
The Characteristics of Pathogenic Autoantibodies
The precise properties of pathogenic antibodies that deposit in the kidneys and elicit an inflammatory response have still not been completely defined. Antibodies eluted from kidneys of both mice and humans with SLE are predominantly class switched, are enriched for anti-DNA activity, and are cross-reactive with multiple autoantigens, including GBM components, whereas lupus serum anti-DNA antibodies are less cross-reactive and “natural” anti-DNA antibodies derived from normal serum have no cross-reactivity.22,23 Some of this polyreactivity may be artifactual, resulting from tight binding of nuclear material to the anti-DNA antibodies, thereby forming a “bridge” to other antigens such as collagen, histones, and glomerular material.24,25 Antibodies eluted from the kidneys also have higher avidity for DNA than those antibodies in the circulation, although there is considerable variability in both compartments.22 Binding to nucleosomal components may be more important than binding to DNA because nucleosomes may become trapped in the kidneys.22 However, binding to chromatin is not the only mechanism for renal deposition, because antibodies without specificity for any nuclear components may also deposit in the kidneys and cause renal damage in murine lupus models.3 Similarly, only a variable proportion of the antibodies eluted from kidneys of patients with SLE are specific for DNA or other known nuclear antigens.23 Another characteristic of pathogenic antibodies is cationic charge, which may confer binding specificity for negatively charged DNA or heparan sulfate (HS) in the GBM. In mice, charged residues such as arginine, lysine, and asparagine in the complementarity-determining regions, particularly of the heavy chain, are associated with anti-DNA–binding activity and pathogenicity.25,26 A number of investigators have reported that anti-DNA antibodies penetrate into live cells, including resident renal cells, via a mechanism dependent on the antigen-binding region of the antibody.27 Although localization of antibodies in nuclei could cause alterations in protein synthesis or impair other nuclear functions, it remains to be shown whether this phenomenon is actually associated with pathogenicity. Studies in vitro have shown that anti-DNA antibodies directly induce proinflammatory cytokine release in mesangial and tubular cells as well as increased proliferation and changes in viability. The mechanisms for these effects are not yet fully elucidated. Finally the isotype of the antibody dictates its effector properties, particularly the ability to bind to Fc receptors and to activate complement (Box 18-1).
Box 18-1
Factors That May Contribute to Pathogenicity of Anti–Double-Stranded DNA Antibodies
1. Functional features associated with antibody deposition:
2. Structural features of the Ig component that influence antigen binding or recruitment of downstream effector pathways:
3. Features of the complexes themselves that affect the amount of renal damage:
Not all anti-DNA antibodies are pathogenic. In one study, infusion of 24 different monoclonal anti-DNA antibodies into mice in vivo resulted in variable patterns of deposition and only some induced pathologic renal changes and proteinuria, indicating heterogeneity in renal specificity.28 Furthermore, the ability to bind to glomeruli in vitro or to induce renal pathology in vivo was found in a second study to be independent of relative avidity for DNA.29 Another comprehensive study of a large panel of monoclonal antibodies derived from SLE-prone mice showed that antibodies to double-stranded DNA (dsDNA) were more pathogenic than antibodies to histones or nonnuclear antigens and that anti-DNA antibodies with glomerular binding specificity were more pathogenic than those that did not bind to glomeruli.25 The abrogation of glomerular binding by DNAse treatment of either the antibodies or the glomerular substrate, with the restoration of binding activity by the addition of chromatin, suggests that much of the glomerular binding of these in vitro glomerular binding monoclonal antibodies is mediated via antigenic bridges.1
To determine whether there are any serum autoantibody specificities that correlate with or predict renal disease in human SLE, clinical studies have compared the specificity of antibodies in the sera of patients with and without nephritis. On a proteome array consisting of multiple autoantigens and renal antigens, serologic reactivity of IgG antibodies with an antigenic cluster that included nuclear antigens and whole glomerular lysates correlated with current renal activity and with overall disease activity as indicated by Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) score.2 As shown for mouse monoclonal antibodies, pretreatment of lupus sera with DNAse reduced binding to glomerular lysates on the proteome array but not to all of the individual glomerular antigens. Another study used the glomerular binding assay to test sera from a well-characterized cohort of patients with SLE with and without nephritis. In this study glomerular binding activity was present in 92% of sera from those individuals with both renal disease and anti-dsDNA antibodies, compared with 25% of sera from individuals with anti-dsDNA antibodies but without nephritis. However, glomerular binding activity did not correlate with either the type or the severity of the renal lesion and was only rarely present in nephritic individuals who did not have anti-dsDNA antibodies in their serum.30 Although these studies in sum show that glomerular binding antibodies are detected more commonly in the sera of patients with nephritis than in those without it and correlate with the presence of anti-DNA antibodies, they also reflect the heterogeneous characteristics of the circulating autoantibodies. Importantly, circulating autoantibodies may not fully reflect the characteristics of those antibodies that actually deposit in the kidneys.
Mechanisms of Tissue Deposition of Immune Complexes
Several overlapping hypotheses have been proposed to explain how autoantibodies deposit in the kidneys (Figure 18-3, A to C).
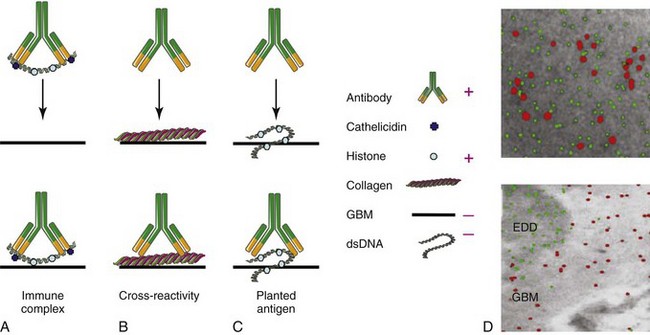
Trapping of Preformed Immune Complexes
The kidney is particularly susceptible to immune complex trapping because it receives a large amount of the cardiac output and has a large glomerular capillary bed. On the basis of findings in mouse lupus models, it was initially thought that circulating immune complexes of antibodies with nucleic acids formed in the blood of patients with SLE become trapped in the mesangium or in the subendothelial space; these are too large to cross into the subepithelial space unless they dissociate. Because small complexes are soluble and do not bind complement efficiently and large complexes are rapidly removed from the circulation by phagocytosis, it has been hypothesized that medium-sized complexes are the most likely to deposit.31 The charge of the complexes may also be important because the GBM is highly negatively charged. Although circulating immune complexes have been detected in animal models of SLE nephritis and have been reported by some investigators in humans with active nephritis, they have been difficult to detect by standard methods.32–33 On the basis of these data and the inability to show that preformed antibody/free DNA complexes deposit in the kidneys, the immune complex–trapping hypothesis was subsequently discredited. However, this hypothesis has now been revisited because it has become clear that a major cause of SLE is a failure to adequately clear apoptotic cell debris and nucleosomes that are rich sources of nucleic acids. Circulating autoantibodies can bind to autoantigens that are exposed in this debris. In fact, nucleosome-complexed antibodies bind more avidly to the GBM than antibodies that have been highly purified and no longer contain nucleosomal material.4 One hypothesis is that binding of positively charged anti-DNA antibodies to the negatively charged DNA of the nucleosome leaves the positively charged nucleosomal histones available to bind to the negatively charged GBM (see Figure 18-3A). This idea is supported by the observation that binding of autoantibodies to the GBM can be blocked by heparin, which acts as an inhibitor of HS binding. In contrast, antihistone antibodies that mask the nucleosomal positive charges prevent binding to the GBM and thus are less pathogenic.24
It has also been shown that circulating microparticles that are released from a variety of cell types are coated with IgG more frequently in plasma from patients with lupus than in plasma from normal controls.34 Patients with SLE also have abnormal neutrophils that are activated in patients with active disease and more prone to cell death via a process called NETosis, in which neutrophils extrude weblike structures, called neutrophil extracellular traps (NETs), that contain large amounts of nuclear DNA. This DNA is complexed with neutrophil-derived antimicrobial proteins such as LL37 (cathelicidin) and human neutrophil peptides belonging to the α-defensin family. Immune complexes containing DNA complexed to these neutrophil peptides have been detected in SLE sera. Not only are these complexes strong inducers of type I interferons (IFNs) but they are also prone to form particulates that are resistant to DNAse treatment.35 Resistance to DNAse can be due to circulating inhibitors or to antibodies that cover the NETs and protect them from the enzyme. Importantly, preliminary studies have suggested that failure to degrade NETs is associated with a higher incidence of nephritis.36
Cross-Reactive Renal Antigens
The second proposed mechanism for tissue deposition is in situ immune complex deposition by direct cross-reactivity with renal antigens other than nucleic acids. The glomerular antigen that is the target in most cases of idiopathic membranous nephritis in humans, phospholipase A2 receptor, has now been identified. However, antibodies to this antigen are only rarely found in cases of membranous SLE nephritis. Nevertheless, many studies have shown that antinuclear antibodies (ANAs) may cross-react with a variety of renal antigens, including laminin, fibronectin, collagen IV, HS, and α-actinin.1 Furthermore, anti-DNA antibodies from patients with SLE nephritis deposit directly in rat glomeruli when perfused into the renal artery, whereas anti-DNA antibodies from patients without nephritis do not.37 Studies of monoclonal antibodies from lupus-prone mice showed that antibodies with cross-reactivities to different glomerular proteins can bind to different types of glomerular cells and may elicit different types of renal diseases.28 One of the problems with using serum antibodies or even monoclonal antibodies for these studies is that they have a strong tendency to bind to circulating nuclear material or to nucleosomes released into the culture supernatants of the hybridoma cells, and it can be very difficult to remove all the nuclear antigens even with DNAse. Thus some of these findings may be explained by the presence of nuclear material bridges. It is also clear that immune complex–mediated renal disease can occur both in SLE-prone mice and in patients without anti-DNA antibodies in their sera; the relevant antigens and mechanisms in the latter remain to be determined.
In Situ Immune Complex Formation
The third major hypothesized mechanism for tissue deposition of autoantibodies is the entrapment of circulating autoantibodies by nucleosomal material (“planted antigen”) that has already bound to renal antigens. Deposition of nuclear material is mediated by binding of positively charged histones to the negatively charged GBM. Relevant GBM antigens include heparan sulfate, collagen type IV, and anionic phospholipids. The source of this material could include circulating chromatin,24 microparticles,34 neutrophil-derived NETs,36 or debris released from locally damaged cells. Further evidence for the planted antigen theory has come from elegant immunohistochemical studies in NZB/W mice showing that in vivo–bound autoantibodies fail to colocalize with the putative cross-reactive antigens, α-actinin, laminin, and collagen IV, but rather colocalize with electron-dense deposits containing chromatin (see Figure 18-3, D).24 Deposition may then be amplified by circulating rheumatoid factors that bind to the Fc region of the antibodies or by antibodies to C1q that bind to fixed complement. Antibodies to C1q may also bind directly to apoptotic material, perhaps through binding to exposed phosphatidyl serine.38 A better understanding of the sources of renal chromatin may lead to new interventions directed at preventing its renal deposition or enhancing its clearance. For example, it has been shown by one group of investigators that SLE nephritic kidneys downregulate their production of DNAse and therefore may be less able to clear planted nuclear antigens.4
Pauci-Immune Glomerulonephritis
Some cases of lupus nephritis are associated with only small amounts of immune deposits. This pauci-immune nephritis may be associated with thrombotic microangiopathy, with vasculitis, or with a podocytopathy that is presumably due to circulating immune mediators that induce activation and damage of intrinsic renal cells.39 In the MRL/lpr mouse model, mild to moderate renal disease can occur even when circulating immunoglobulins are absent, suggesting that innate and T cell–mediated mechanisms may be sufficient to cause tissue damage.9 Several new studies have investigated the role of cell-mediated immunity in the induction of renal damage. There is evidence that interstitial renal dendritic cells, when activated by renal injury, can capture renal antigens that have been degraded and transported from the tubular lumen by tubular epithelial cells. Because self-antigens constitute the majority of peptides expressed in the peptide-binding groove of MHC class I molecules, renal cells may then become targets of activated cytotoxic T cells.40
Mouse Models of Lupus Nephritis
Two main categories of mouse models exist, namely, spontaneous models41 and induced models. Not surprisingly, striking differences in both pathogenic mechanisms and responses to immunologic interventions have been observed in the different mouse models, suggesting that multiple animal models will be needed to dissect the pathogenetic mechanisms of SLE nephritis and to study responses to new therapies. These differences among the mouse models parallel the emerging appreciation of heterogeneity in human SLE nephritis.42 Apart from the limited heterogeneity of the mouse models, a further limitation of the mouse in comparison with human and other species such as rat is the relative resistance to the development of end-stage kidney disease, most likely as a consequence of a higher nephron endowment relative to body mass than in other species. However, given the vast literature on mouse models of SLE on which potential new therapies are often based, this chapter contains a brief description of several murine models in which the renal disease has been well characterized.
Diffuse Proliferative Glomerulonephritis with Anti-DNA Antibodies
The NZB/W F1 hybrid, the oldest classic SLE model, has a phenotype comparable to that of patients with lupus, with the production of IgG2a anti-dsDNA antibodies and some genetic similarities to human SLE. Nephritic kidneys are characterized by proliferative glomerulonephritis with glomerular enlargement and hypercellularity, extensive infiltration with activated renal macrophages in the tubulointerstitial region, and accumulation of T and B cells along with dendritic cells in disorganized perivascular and periglomerular aggregates,43 similar to those reported in SLE biopsy specimens. These mice can also be used to study disease remission and relapse because complete remission of established nephritis is achieved in more than 80% of mice by combination therapy either with a short course of cyclophosphamide (Cytoxan) together with co-stimulatory blockade or with a short course of cytotoxic T-lymphocyte antigen 4 (CTLA-4) Ig together with B-cell activating factor (BAFF) blockade.44 A similar hybrid is the SNF1 mouse, a hybrid of SWR with NZB. This mouse demonstrates proliferative disease with glomerular hypercellularity with crescents, thickening of capillary loops and basement membrane as well as mesangial thickening, large perivascular infiltrates of lymphoid cells, and eventually, glomerulosclerosis and fibrosis.45
Sclerotic Glomerulonephritis with Antinucleosome and Anti-DNA Antibodies
The NZM2410 strain of mice was derived from a back-cross between NZB/W F1 and NZW mice followed by brother-sister mating. Twenty-seven different recombinant inbred strains of New Zealand Mixed (NZM) mice were obtained; among them, NZM2410 and NZM2328 have been used most commonly as lupus models. NZM2410 mice are dominated by an interleukin-4 (IL-4) response, and they produce IgG1 and IgE antichromatin antibodies. They demonstrate sclerotic glomerulonephritis with early loss of podocytes, and they accumulate activated interstitial macrophages but have little lymphocytic or dendritic cell infiltration.46 These mice are very responsive to treatment, and BAFF blockade alone induces long-term survival and may even induce remission of established nephritis.47 A congenic strain of C57BL/6 mice named Sle1,2,3 has been generated with the use of three genetic loci from the NZM2410 mouse.
NZM2328 Mice and Congenic Strains—Proliferative Disease without Antinuclear Antibodies
NZM2328 are phenotypically similar to NZB/W and have been used extensively to study the effects of single-gene deletions on the immune system. An important finding in this system is that deficiency of signal transducer and activator of transcription 4 (STAT4), which decreases T-helper 1 cell (Th1) cytokine responses, results in accelerated nephritis despite a marked decrease in serum IgG2a autoantibodies, whereas deficiency of STAT6, which decreases Th2 cytokine responses, ameliorates nephritis despite the presence of high-titer serum IgG2a autoantibodies.48 Both STAT4- and STAT6-deficient mice have less renal immune complex deposition than wt NZM2328 mice, indicating the complex relationships between autoantibody specificity, autoantibody deposition, and renal pathology. NZM2328 congenic mice have been derived by back-crossing to C57L. One of these strains demonstrates immune complex–mediated glomerulonephritis but has no ANAs or rheumatoid factor activity in either the serum or renal eluates.3 The relevant renal antigens have not yet been identified.
Proliferative Disease Associated with Anti-RNA and Antiphospholipid Antibodies
BXSB.Yaa mice exhibit monocytosis and lymphoid hyperplasia, circulating immune complexes, and proliferative immune complex glomerulonephritis. Male BXSB mice carry the Yaa (Y chromosome–linked autoimmunity accelerator) locus, consisting of a reduplication of at least 17 genes from the X chromosome, including TLR7; the resulting dysregulation of the innate immune response induces the development of antibodies to RNA antigens and accelerates disease onset.49 NZW/BXSB F1 (W/B) mice produce anti-Sm/RNP (ribonucleoprotein) antibodies and antiphospholipid antibodies, which cause clots within the myocardial small vessels. These mice have severe proliferative glomerulonephritis, with disorganized large lymphoid infiltrates, scattered interstitial T cells, sheets of macrophages in the interstitium and periglomerular regions, renal vasculitis, and the accumulation of dendritic cells within the glomeruli.50 Serologically active W/B mice are resistant to therapy with cyclophosphamide and co-stimulatory blockade but have a partial response to treatment with BAFF blockade.50 The discovery of the TLR7 gene within the Yaa locus has led to the development of a TLR7 transgenic mouse that is on a nonautoimmune genetic background; even females carrying more than four copies of TLR7 spontaneously demonstrate SLE with features similar to those in the BXSB mouse.
Proliferative and Interstitial Disease Associated with Anti-DNA, Anti-RNA, and Rheumatoid Factor Autoantibodies
MRL/lpr mice have a permissive genetic background and are also deficient in the proapoptotic molecule Fas, resulting in accumulation of double-negative (CD4−/CD8−) T cells and B220+ cells51 as well as massive lymphoproliferation. These mice have elevations of multiple autoantibodies, including ANA, anti–single-stranded DNA, anti-Sm, and rheumatoid factors, and make large quantities of IFN-γ and IgG2a autoantibodies.46 They demonstrate diffuse proliferative glomerulonephritis with mesangial cell proliferation and crescent formation as well as significant interstitial nephritis; mild glomerular disease and progressive interstitial disease can occur even in the total absence of circulating autoantibodies.9 Macrophage infiltration is an essential feature of nephritis in this model and is driven by renal expression of colony-stimulating factor 1 (CSF-1).52 The infiltrating cells have not as yet been well characterized in this model, although recruitment of F4/80lo/Ly6Chi inflammatory macrophages into the kidneys is observed 1 or 2 days after adoptive transfer of bone marrow–derived monocytes.52 When MRL/lpr mice are rendered deficient in the IL-27 receptor, their autoantibody response is skewed to a Th2 phenotype, they express high levels of IL-4, IgG1, and IgE,53 and membranous nephropathy develops; these mice have longer renal survival than their IL-27R–sufficient counterparts. These findings indicate that even in mice with the same genetic susceptibility to SLE, the cytokine milieu can have a major impact on the type of nephritis that develops.
Nephrotoxic Nephritis
Acute immune complex–mediated glomerulonephritis is induced in rats and in certain strains of mice by infusion of sheep or rabbit antibodies specific for glomerular type IV collagen followed by disease induction with an innate immune trigger such as lipopolysaccharide (LPS).54 The disease is characterized by acute neutrophil and inflammatory Gr1hi macrophage infiltration and T-cell infiltration occurring over days to weeks. The acute phase is followed by a period of renal repair and recovery. Much information is available about the mechanisms of tissue damage in this acute model, which has been invaluable in defining the cells and mediators necessary for acute renal inflammation. Both Fc receptors and complement activation are required, as are the acute-phase cytokines TNF-α, IL-1, and IL-6. The T cells involved appear to be skewed to a Th1 phenotype, and PDGF, a molecule that enhances mesangial cell proliferation and matrix formation, is also required. In addition, the observation that only some strains of mice are susceptible to disease induction led to elegant genetic studies in which polymorphisms of the kallikrein genes were identified as major determinants of disease susceptibility.55 The nephrotoxic nephritis model has also been useful for identifying both the pathogenic role of inflammatory Gr1hi macrophages during the early phases of disease and the protective role of reparative macrophages during disease resolution. However, this disease has many differences from spontaneous SLE models, in which neutrophils and inflammatory Gr1hi macrophages are infrequent. Thus, although it is a useful model for studying the effector phase of acute inflammation, it is not adequate for the study of chronic SLE nephritis.
Pristane-Induced Nephritis
The hydrocarbon pristane can induce a lupus-like disease in several strains of mice with the induction of autoantibodies against DNA, chromatin, Sm, ribonucleoprotein, and ribosomal P. These mice demonstrate immune complex–mediated proliferative glomerulonephritis with mesangial and subendothelial deposits. Interestingly, although the absence of type I IFN signaling prevents the formation of antibodies to DNA and RNA, antinuclear and anticytoplasmic antibodies are still found in the serum, and IgG and complement deposition in the kidneys still occurs. Nevertheless the immune complexes do not induce renal damage, indicating that type I IFN signaling is required for the subsequent effector stage of disease.56
Kidney Effector Mechanisms
Although immune complex deposition in the kidneys is a critical pathogenic component in most cases of SLE nephritis, it is not sufficient to cause inflammatory renal disease that injures the kidney. Multiple studies in mice have identified key pathways that are required for renal damage to occur. Immune complex deposition without renal damage has been reported in SLE-prone mice deficient in FcR γ-chain,57 IFN-α/β receptor (IFNAR),56 or monocyte chemotactic protein 1 (MCP-1),58 and in mice treated with caspase inhibitors, total lymphoid irradiation, combination cyclophosphamide/co-stimulatory blockade,43 or very high doses of anti-CD154 antibodies. These findings are consistent with a disease model in which immune complex deposition in the kidney triggers a cascade of inflammatory events mediated by activation of complement or engagement of FcRs on renal cells or circulating monocytes, followed by upregulation of renal and monocyte-derived inflammatory chemokines; transmigration of inflammatory cells into the renal parenchyma; release of damaged tissue, inflammatory cytokines, and mediators that amplify the process; and, finally, irreversible renal damage. This model suggests many avenues for therapeutic intervention that are not necessarily based on systemic immune suppression (Figure 18-4).
Complement
Renal deposition of autoantibodies of the appropriate isotype is followed by recruitment of complement components.5 The complement system comprises more than 30 proteins, some of which are activating and some of which are regulatory. Complement plays an important role in clearance of apoptotic material and immune complexes, and therefore, early component complement deficiencies that cause an overload of circulating nuclear material are highly associated with susceptibility to SLE in humans. In contrast, excessive activation of later complement components may be associated with failure to control inflammation or thrombosis; several renal diseases have been associated with loss-of-function mutations in complement regulatory proteins.
Activation of complement may occur through the classical pathway, which is activated by the binding of C1 to immunoglobulins in immune complexes, or the lectin pathway, which is activated by the binding of mannose-binding lectin to terminal carbohydrates on microbes. A third, “alternative” pathway is an amplification pathway that is activated by binding of hydrolyzed complement factors 3b and 3d, generated via the other two pathways, to factor B, resulting in formation of an alternative C3 convertase (Figure 18-5). An increase in synthesis of various complement components may occur at sites of inflammation or hypoxia; for example, IL-1, IL-6, IFN-γ, and TNF-α all upregulate C3 production. Studies in MRL/lpr SLE-prone mice have demonstrated that deficiency of factor B, an essential component of the alternative pathway, protects from renal disease, whereas deficiency of C3, which is required both for complement activation and for clearance of unwanted material, does not confer protection.59,60
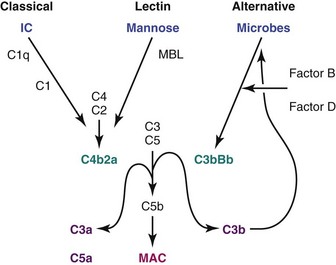
Cleavage of complement components C3 and C5 by the complement convertases that are generated via the three complement activation pathways amplifies complement activation (C3b), releases proinflammatory products (C3a and C5a) that cause tissue damage through anaphylotoxin activity, and activates the terminal complement membrane attack complex (MAC C5b,C6-9). The MAC has both cytolytic and noncytolytic effects. Activation of the MAC in membranous disease results in podocyte injury, which disrupts the actin cytoskeleton and releases oxidants and proteases that damage the GBM. The MAC also induces proliferation of mesangial cells and stimulates their release of profibrotic mediators TGF-β and PDGF. Finally the terminal complement products can activate endothelial cells, inducing the expression of adhesion molecules and other inflammatory mediators and acting as procoagulants.61
Targeting of the effector molecules of the complement pathways, including C5 and the various regulatory proteins, may be therapeutic in renal inflammation. For example, a therapeutic agent that targets the soluble complement regulatory protein factor H to renal tubular sites of C3 binding has shown some efficacy in mouse models of renal ischemia.62 Eculizumab, the only currently approved complement inhibitor, is a humanized monoclonal antibody that prevents the cleavage of human complement component C5 into its proinflammatory components. This agent is therapeutic in diseases associated with deficiency of complement regulatory proteins, such as hemolytic uremic syndrome, and is currently in clinical trials for several chronic inflammatory diseases, including nephritis.61
Another complement-based amplification mechanism is mediated by antibodies to tissue-bound C1q, which is the first component of the classical pathway and binds directly to apoptotic cells and to aggregated immunoglobulin, particularly IgG3. C1q binding causes a conformational change in the molecule that may render it immunogenic; thus, defective clearance mechanisms in SLE may predispose to the generation of anti-C1q autoantibodies in a fashion analogous to the generation of antibodies directed to nucleic acids. High titers of anti-C1q antibodies, particularly of the IgG2 isotype, have been associated with lupus nephritis in multiple studies of patients with SLE. When anti-C1q antibodies are infused into the kidneys of normal mice, they recruit C1q but do not cause renal injury. However when anti-C1q is infused into the kidneys of mice with low levels of immune complex deposition, they can trigger renal inflammation; this finding suggests that the antibodies bind to or recruit C1q onto immune complexes, thereby activating the classical complement pathway. C1q may also be recruited to damaged cells in the kidney that then act as targets for anti-C1q antibodies.38,63 Alternatively, the antibodies may interfere with complement-mediated clearance of immune complexes and apoptotic material.
Related posts:
 The Innate Immune System in SLE
Clinical Aspects of the Antiphospholipid Syndrome
Gastrointestinal and Hepatic Manifestations
Novel Therapies for SLE: Biological Agents Available in Practice Today
Mixed Connective Tissue Disease and Undifferentiated Connective Tissue Disease
Pathomechanisms of Cutaneous Lupus Erythematosus
The Innate Immune System in SLE
Clinical Aspects of the Antiphospholipid Syndrome
Gastrointestinal and Hepatic Manifestations
Novel Therapies for SLE: Biological Agents Available in Practice Today
Mixed Connective Tissue Disease and Undifferentiated Connective Tissue Disease
Pathomechanisms of Cutaneous Lupus Erythematosus
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


