Fig. 32.1
(a) Notice the left-sided acetabulum and proximal femur regions’ bone marrow edema (fat-suppressed T2-weighted coronal image). This scan was obtained at the beginning of bisphosphonate treatment. (b) Three months after the bisphosphonate treatment of the patient, left-sided acetabulum and bilateral proximal femoral regions’ bone marrow edema was apparently subsided (resolved)
A 29-year-old man admitted to our outpatient clinic with bilateral hip pain and limp which was prominent left side. One year before he underwent cortisol treatment after the intracranial surgery due to situation of intracerebral hemorrhage which caused by intracerebral vascular malformation. The patients’ complaints have started 1 month before the admission to our outpatient clinic. The physical examination revealed limited range of motion at flexion and rotations of the hips. The Trendelenburg test was also positive. MRI fat-suppressed T2-weighted coronal images were consistent with bone marrow edema at left acetabulum and bilateral proximal femoral region.The patient was diagnosed as cortisol-induced osteonecrosis under the lights of MRI imagining findings and well-documented anamnesis. The weekly 70 mg alendronate/oral and vitamin D3-calcium (oral way) treatment were initiated. The nonweight bearing with the crutches (only finger touch was allowed) was recommended. The patient uneventfully healed with the evidence of nonpainful hips and resolution of limited range of motion. After the 3-month treatment with bisphosphonates, the control MRI revealed that even if the total resolution of the bone marrow edema does not exist, a significant amount of edema was subsided especially at the acetabulum and at both hips and after the treatment. The regions of avascular necrosis were also precipitated. After this clinical and radiological advancement of the patient, 1-year bisphosphonate treatment was considered.
Avascular necrosis of the femoral head(ANFH) can cause structural failure of bone with collapse and dysfunction of the hip. Collapse of the femoral head was observed in 75 % of cases of avascular necrosis of the femoral head within 3 years [16]. According to Aaron et al., more than 50 % of patients with avascular necrosis of the femoral head require THR within 3 years of diagnosis [17].
Statins [18], anticoagulants [19], prostacyclin [20], and bisphosphonates (Bps) [21] have been used to treat osteonecrosis of the hip. The theoretical benefit of statins is based on the association of increased fat cell size with an increased risk of the development of osteonecrosis of the hip. Anticoagulants inhibit the aggregation of platelets and enhance blood flow to ischemic areas of bone. Prostacyclin promotes bone regeneration on a cellular or systemic level but fail to show efficacy in the advanced stages of ANFH. Bps are potent antireabsorptive agents that act by inhibiting the action of mature osteoclasts in the bone, which theoretically normalized the uncoupled bone remodeling contributing to femoral head collapse.
Agarwala et al. first reported the efficacy of alendronate, a bisphosphonate, in the treatment of avascular necrosis of the femoral head and showed that it not only improved the symptoms but also retarded progression of the disease and reduced the rate of collapse of the femoral head [22]. Other studies have shown similar results and bisphosphonates are now considered to be one of the standard options of treatment for avascular necrosis of the femoral head [23]. Favorable long-term results were also presented by Agarwala’s 10-year study in treated patients even after alendronate discontinuation [24]. In addition, there were no severe adverse effects associated with alendronate treatment observed during short- or long-term follow-up. Another finding of the current review is that although patients in all stages appeared to have potential benefit from alendronate treatment, the application in early AVN with small-size lesion was suggested by most of the included studies. Specially, as shown in Chen’s study, when extensive osteonecrosis (stages II C and III C) was radiographically presented, alendronate did not have any benefits. Thus, the efficacy of alendronate for AVN with large necrotic legions should be considered more carefully [25].
Osteonecrosis
Osteonecrosis (ON) is the ischemic necrosis of the bone and bone marrow. Ischemia only affects the cells and not the organic or inorganic matrix. It might develop in every bone and every part of the bone. Clinical status is determined by the ischemic location on the bone. While an ischemic island on a spongious bone is symptom-free, a necrosis toward subchondral bone (such as femur head avascular necrosis) shows quite dramatic clinical features.
Etiopathogenesis
ON is usually caused by the disturbed blood flow in the bone due to vascular trauma or vascular embolism. The main causes can be summarized by the mnemonic “VINDICATE” (Table. 32.1).
Table 32.1
Causes of avascular necrosis
Causes | |
|---|---|
V | Vascular |
IN | Infection |
D | Drugs |
I | Inflammation |
C | Congenital |
A | Autoimmune |
T | Trauma |
E | Endocrine |
Most frequent is the association with prolonged corticosteroid therapy or excessive alcohol intake. Other risk factors include hyperlipidemias, coagulopathies (thrombophilia, hypofibrinolysis), abnormal red blood cells (sickle cell), dysbarism, HIV infection, smoking, and genetic factors [1, 2]. Many etiological factors are responsible for vascular damage. These mechanisms are mechanical interruption, thrombotic occlusion, or extravascular compression (Fig. 32.2).
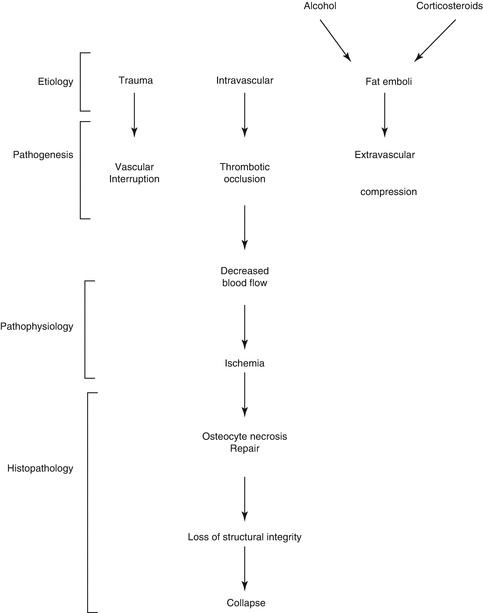
Fig. 32.2
Many etiological factors are responsible for vascular damage. These mechanisms are mechanical interruption, thrombotic occlusion, or extravascular compression [3]
Factors for Pathogenesis
1.
Idiopathic (bilateral in 40–50 %): Most of the time, there is no clear reason for pathogenesis. Coagulation anomalies might be the cause in this group.
2.
Arterial tears: In dislocations and fractured bones
3.
Arteriolar embolism: Embolism, sickle-cell crisis, and decompression sickness (caisson disease)
4.
Vascular wall damage: Vasculitis (SLE) and radiation
5.
Capillary embolism due to fat tissue infiltration of bone marrow:
(a)
Steroid treatment (>30 mg prednisolone, >30 days, 37 % risk, 80 % bilateral)
(b)
Heavy alcohol abuse (20 %)
(c)
Gaucher’s disease (Gaucher’s macrophages filling the bone marrow space)
(d)
Pancreatitis
(e)
Hyperlipidemia
(f)
Others: Renal transplant, diabetes, sepsis, inflammatory bowel disease, and hematological diseases
To sum up, in 40 % of the cases, the pathogenesis is unknown, 30 % are caused by cortisone usage, and 20 % by alcohol abuse and 10 % have other reasons.
Pathology
Necrosis happens when the bone tissue is not properly nourished. This does not affect the organic-inorganic matrix. The hematopoietic cells are most sensitive to anoxia. Bone marrow and osteocyte necrosis develops after 2–3 h of ischemia and is permanent [3]. Histological differences can be seen only after 24–72 h (Fig. 32.3).
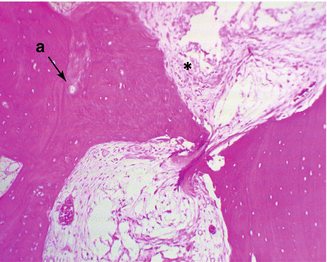
Fig. 32.3
Early stage of osteonecrosis, a. empty lacunas due to osteocyte death, *necrotic bone marrow (Courtesy of Gürer İ MD)
Repair of the Dead Bone
An inflammatory response develops against dead bone marrow. This is followed by chemotaxis and arrival of the mesenchymal cells with vascular generation to repair zone. In the literature, resorption of the dead bone tissue and new bone formation is named as “creeping substitution.” The repair period is determined by the necrotic area size and whether the necrotic bone is spongious or cortical type. In spongious bones, this situation can be fixed by the new bone tissue covering the dead bone tissue directly. However, in cortical bones, this happens with a mechanism named “osteoclast cutting cone” (Fig. 32.4). In this mechanism, osteoclasts get into the dead bone with vascular extensions, removing the dead bone tissue, meanwhile mesenchymal cell-developed osteoblasts fill the area with new bone tissue. In direct radiological viewing studies, early ON can be seen as a local density decrease in the necrotic areas. The previous density levels are achieved after the bone healing in 12–18 months. MRI studies however show the bone marrow edema as hypointense in T1 and hyperintense signals in T2 sequence.
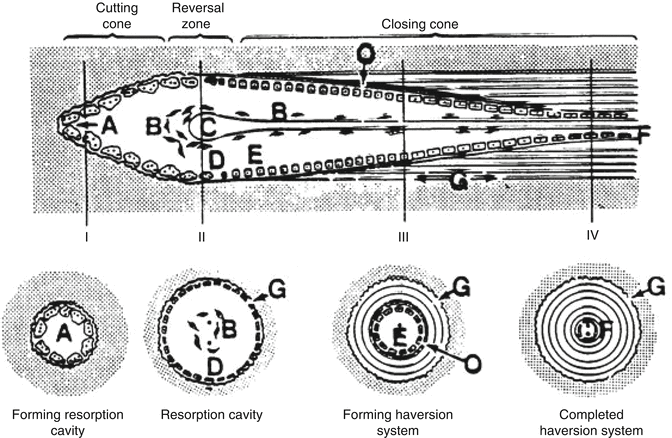
Fig. 32.4
Osteoclast cutting cone in repairing and reshaping the dead bone. Osteoclasts bore the dead matrix like a tunnel-boring device in front of a vascular extension and osteoblasts cover the surface with bone tissue. This is called “creeping substitution” (http://www.mobileappscentre.co.uk/healthapp/author/admin/)
Dead bone tissue has lost its living cells and vitality. Since there are no living cells present, bone turnover cycle does not happen in necrotic area. For this reason, stress fractures or necrotic bone collapse can be seen with physiological stress in the necrotic area. Those changes are especially important in subchondral bones. The collapse and destruction of subchondral bone (crescent sign) (Fig. 32.5) disturb the integrity of the joint such as hip joint and causes early arthrosis. If the subchondral bone is protected from the stress, microfractures, and collapses, the joint can be saved by the repair of dead bone tissue. The repair tissue that covers the subchondral bone (Fig. 32.6) can sometimes inhibit the vascular entry to the bone [4]. In osteonecrosis of femoral head (ONFH), multiple drilling and vascular-avascular fibular grafts are used to facilitate the vascular entry to dead zone and prevent collapses.
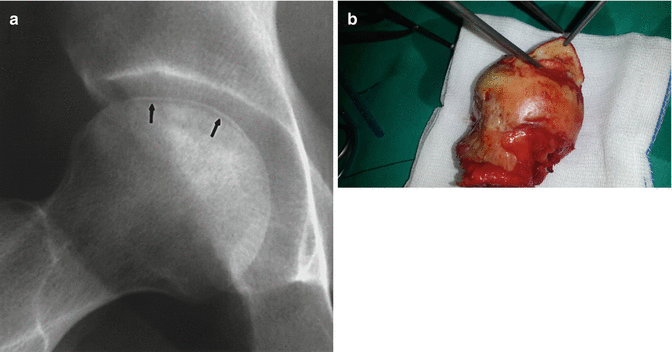
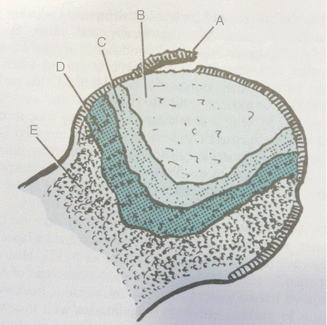
Fig. 32.5
Separation of subchondral bone (crescent sign). (a) X-ray (arrows) and pens show the separation of subchondral bone. (b) Intraoperative photo
Fig. 32.6
Avascular necrosis pathology. A Joint cartilage and subchondral separation. B Necrotic bone. C Reactive connective tissue surrounding dead tissue (keeps vascular tissue out of dead bone zone). D Reactive bone tissue that limits the dead bone. E Normal bone tissue (Miller [4])
Clinical Features and Imaging
Clinical symptoms and features show the location and area width of the necrosis. Small, metaphyseal area and spongious bone-located necrosis shows no symptoms, yet necrosis that extends toward subchondral bone shows clear features. If the area of involvement is small (<25 % of the femoral head) and if it is not close to the articular surface, the repair process may be sufficient to maintain the structural integrity of bone. Small medial lesion of femoral head has an especially good prognosis [1].
Imaging studies can show the different stages of osteonecrosis (early stage, fragmentation, repair, and late) by changes in density, subchondral separation and fragmentation, repair, and arthrosis development. Radiographic studies of avascular necrosis in the early stages usually appear normal. The earliest radiographic sign of osteonecrosis is the presence of radiolucent crescent (the so-called crescent sign) (Fig. 32.5a). In the later stages, it appears relatively sclerotic due to nearby living bone becoming low density secondary to reactive hyperemia. Ficat and Arlet [5] described the first staging system. They described four stages based on plain radiographs of the affected hip. Stage I indicated a clinical suspicion of ONFH with no changes noted on X-ray films. In stage II there are changes, including osteosclerosis and/or cysts with conservation of the normal contour of the femoral head. The crescent sign characterizes stage III, which is seen as subchondral collapse or flattening of the femoral head on plain radiographs. Stage IV consists of narrowing of the joint space and secondary degenerative changes within the acetabulum.
MRI of osteonecrosis can show changes earlier than radiography or CT. The sensitivity and specificity of MRI are also very high. MRI can show the ischemic bone marrow changes faster than scintigraphy. Ischemic area shows itself as hypointense signals in T1-weighted images (Fig. 32.7a) and hyperintense in T2-weighted images (Fig. 32.7b) due to bone marrow edema. When present, the double-line sign on the T2-weighted image is essentially pathognomonic for osteonecrosis [1].
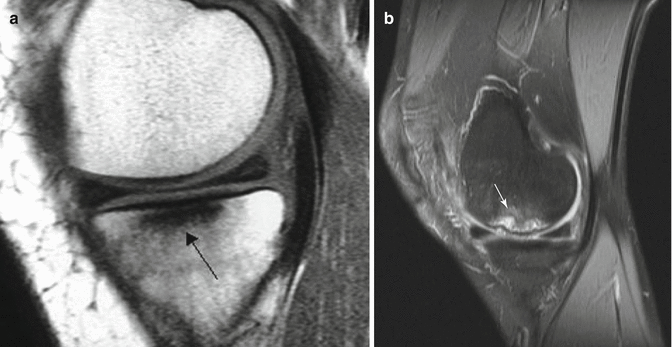
Fig. 32.7
(a) T1 sequence shows osteonecrosis area as hypointense (arrow). (b) T2 sequence shows osteonecrosis area as hyperintense (white arrow)
Metabolic Bone Disorders
Metabolic bone diseases are systemic diseases that affect the bone development, mineralization, and remodeling. They make the bone easily breakable and deformable by causing disruptions in bone mass, composition, and remodeling.
Bone mass is the weight of the bone unit volume. It is not possible to weigh the bone in vivo. Bone mass is determined by bone mineral density measurements and changes in the radiological density of the bone (osteopenia, osteosclerosis). Bone mass is mainly affected by osteoblastic activity and the changes in bone turnover cycle. The most important metabolic disease that affects bone mass is osteoporosis. In osteoporosis, bone mass is reduced and bones are easily breakable.
Bone composition is the rate of mineralized and demineralized matrix (Fig. 32.8). This is directly connected to the matrix mineralization. Metabolic bone diseases affect the mineral metabolism, therefore changing bone composition. In rickets-osteomalacia syndromes, demineralized bone matrix rate is increased and bone composition is changed. In this disease group, the bones become softer since the bone matrix is not properly mineralized and could be easily deformed without breaking.
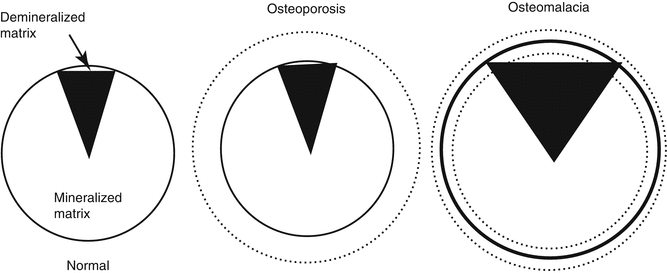
Fig. 32.8
Composition of the bone. In osteoporosis, there is a loss of mass without the composition change. On osteomalacia, the mass can either increase or decrease. The main change is the increased demineralized matrix amount (From Netter [6])
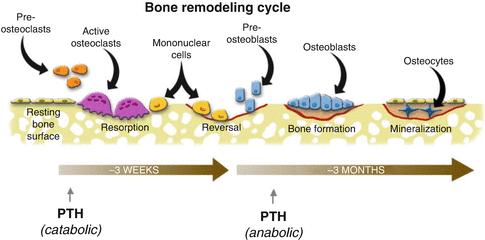
Bone remodeling is controlled by the bone remodeling cycle (Fig. 32.9). Bone remodeling cycle is one of the most important homeostasis mechanisms of the body. Development and persistency of the bone are regulated by this mechanism in addition to fracture healing, repair of the dead bone areas and grafts. The most typical diseases which cause a disruption in the local and wide bone remodeling mechanism are Paget’s disease and osteopetrosis. In both diseases, bone mechanic properties were changed and bone can be easily fractured and deformed.
Cellular Biology [7]
The development and change of bone mass are by this mechanism. In bone remodeling cycle, osteoblasts, osteoclasts, and osteocytes play an active role.
Osteoblasts synthesize the osteoid (type I collagen) tissue which is bone matrix. They are the primary bone-building cells and develop from mesenchymal stem cell. They synthesize the osteoid matrix and control the mineralization of bone matrix in addition to calcium-phosphate balance of the body. They also secrete osteocalcin and alkaline phosphatase. By activation of osteoclasts, they are also responsible for the bone resorption and regulation. They contain parathyroid, thyroid, and growth hormone receptors in addition to estrogen, androgen, and vitamin D. They can produce local cytokines, growth hormones (IGF, PDGF, bFGF, TGF-b), and bone morphogenetic factor (BMP).
Osteoclasts are responsible for bone resorption. They differentiate from hematopoietic stem cells and develop from monocytes. Osteoblasts play an active role in osteoclast activation. They have receptors for calcitonin, colchicine, and gamma-interferon receptors and answer directly.
Osteocytes are differentiated from osteoblasts and found in lacunas within the matrix. They cannot move and they connect either to each other or toward bone surface by cytoplasm extensions. They have a high nucleus/cytoplasm rate. Their function is not yet fully understood. They are thought to play a role in the transfer of mechanical signals to the bone.
Bone Remodeling Cycle
Bone remodeling cycle starts with osteoclastic activity and bone resorption (Fig. 32.9). Osteoclasts are found in Howship’s lacunas on bone resorption areas. Osteoclasts are synthesized from hematopoietic stem cells and differentiated from monocytes by the secretion of RANKL (receptor activator NF-κβ ligand) protein from osteoblastic stromal cells due to environmental triggers (BMP, 1,25-dihydroxyvitamin D, and PTH). This protein attaches to the RANK protein on osteoclast progenitor cell to differentiate the cell to osteoclasts (Fig. 32.10). Osteoprotegerin (OPG) secreted from stromal cells inhibits this pathway by connecting to RANKL. PTH, PG-E2, and 1,25-dihydroxyvitamin D stimulate the osteoclast formation. Macrophage colony-stimulating factor (M-CSF), which is a cytokine secreted from stromal cells and osteoblasts, connects with c- fms on osteoclast progenitor cell membrane to stimulate the reproduction and fusion of those cells.
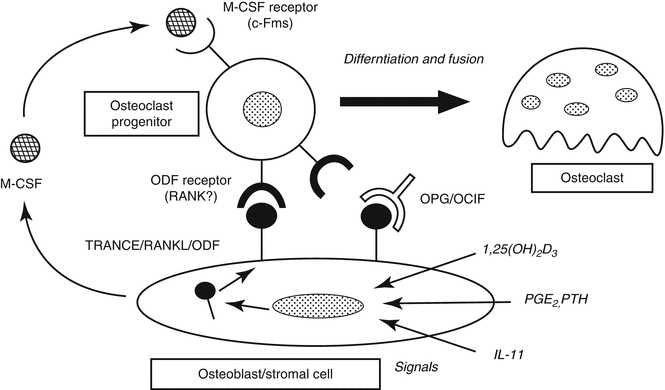
Fig. 32.10
Osteoclastogenesis. Stromal cells differentiate to osteoclast precursors to osteoclast by RANK/RANKL system. Osteoprotegerin (OPG) inhibits osteoclastogenesis by blocking this mechanism. RANK/RANKL reseptor activator of nuclear factor-kappa B ligand, ODF osteoclast differentiation factor, OCIF osteoclastogenesis inhibitory factor, M-CSF macrophage colony-stimulating factor
The structural integrity and mass are controlled by bone turnover cycle. Systemic and local control mechanisms are responsible for this cycle’s continuity.
Systemic factors: Parathyroid hormone (PTH) is the most important hormone in turnover cycles in many of the target organs. It also plays an important role in calcium metabolism by stimulating osteoclastic resorption, increasing calcium resorption from renal tubules, and stimulating calcitriol synthesis in renal parenchyma. If PTH is given constantly, the bone resorption, if it is given intermittent bone development, is stimulated. Calcitriol (Vitamin D3) has an anabolic effect. Calcitonin inhibits the osteoclasts according to dosage to decrease bone resorption. During growth phase, growth hormones (GH, IGF-I and II) enable the growth (increase in bone mass) by their effect on endochondral ossification, growth plates, and bone turnover cycle. Thyroid hormones and glucocorticoids have both stimulating and inhibiting effects on bone cells. The most important hormones that show anabolic effect on the bones are the sex hormones. Estrogen stimulates osteoblast replication while desensitizing osteoclast progenitor cells toward RANKL, therefore limiting osteoclast production. This saves the bone mass. With menopause, this system ends and postmenopausal osteoporosis develops.
Local factors: Local factors affecting the bone turnover cycle in bone cells are the interactions between OPG and RANKL and RANK systems. RANKL/RANK system controls the change from osteoclast primary cells to osteoclasts while OPG totally inhibits this system. Systemic (hormones) and local (cytokines) factors both affect this cycle by “intersection hypothesis.” TNF-alpha and IL-10 cytokine groups increase the osteoclastic function by stimulating m-CSF and RANKL production. This mechanism is effective on bone resorption against prostheses.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


