Chapter 31 Ocular, Aural, and Oral Manifestations
Systemic Lupus Erythematosus and the Eye
The most common of the ocular manifestations is dry eye or keratoconjunctivitis sicca (KCS) as a result of secondary Sjögren syndrome (Chapter 32 discusses this topic in detail). Of the other ocular pathologic conditions, retinal vasculopathy in the form of cotton wool spots is the next most common and has ominous systemic implications. Optic neuropathy, although rare, is associated with a poor visual prognosis. Less common manifestations are also briefly discussed in this section. In addition, this chapter discusses the known side effects of antimalarial drugs—a class of common medications used in SLE.
Retinal Vascular Disease
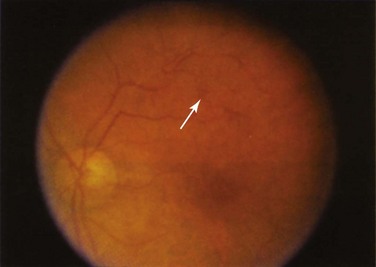
Retinal vascular lesions appearing as localized retinal infarctions at the level of the retinal nerve fiber layer are the most common intraocular manifestations of SLE. These are visible on ophthalmoscopic images as cotton wool spots (Figure 31-1) and are often asymptomatic if located in the periphery of the retina. Occasionally, these cotton wool spots may be associated with intraretinal hemorrhages and may result in a Roth spot (Figure 31-2).
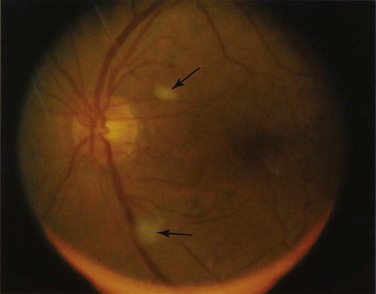
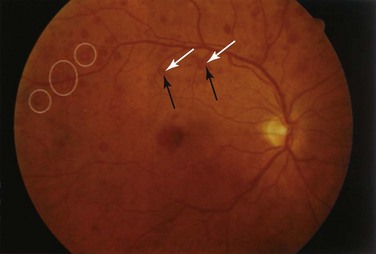
The published prevalence of retinal cotton wool spots in patients with SLE varies from 3% to 29%.1–3 However, the largest prospective study4 with more than 15 years of serial observation of 550 patients with SLE found that these lesions were present in 7% of patients with lupus.
Severe, occlusive retinal vasculopathy is far less common but is usually visually devastating, with one series reporting a final acuity of worse than 20/200 (i.e., legal blindness) in 55% of eyes affected by this type of disease.2 The manifestations seen in this type of vasoocclusive disease include central retinal artery occlusions (Figure 31-3), multifocal retinal arteriolar occlusions (Figure 31-4), capillary bed occlusion resulting in widespread retinal ischemia with secondary retinal and optic disc neovascularization that may lead to vitreal hemorrhage (Figure 31-5) or tractional retinal detachment, and central and branch retinal vein occlusions (Figure 31-6).2,4–9 Patients with any of these occlusions usually complain of a sudden, painless loss of vision or a loss of visual field or both that classically respects the horizontal meridian. Fortunately, this subtype of retinal vasculopathy is rare with an incidence of less than 1% in the previously mentioned prospective series of 550 patients.4 Other smaller series have reported an incidence of 2% to 8%.1,10
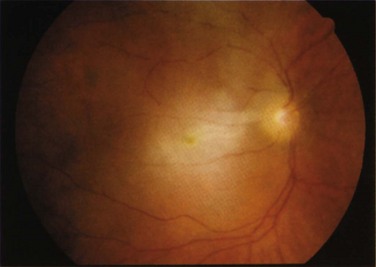
FIGURE 31-3 Central retinal artery occlusion of the right eye. The ischemic, pale, edematous retina is evident.
Although retinal vascular involvement is most commonly asymptomatic in patients with SLE, its presence is associated with active SLE in 88% of patients and with lupus cerebritis in 73% of patients.2,4 A strong correlation between the presence of retinal vascular involvement and lupus anticoagulant or antiphospholipid antibodies has also been found in several studies in addition to a decreased survival time.2,4,5,10,11 The presence of lupus retinal vascular disease is therefore a marker of poor prognosis for survival.
Choroidal Vascular Disease
Retinal vessels are readily seen with an ophthalmoscope; therefore retinal vascular disease is relatively easy to assess. In contrast, the choroidal vasculature is deep to the retina and usually obscured by the retinal pigment epithelium. Abnormalities of choroidal vessels in SLE are rarely reported but are also significantly more difficult to recognize than retinal vascular disease. A choroidal vasculopathy usually results in multiple foci of serous retinal detachments that eventually resolve with residual scarring of the retinal pigment epithelium and some degree of permanent visual impairment.5,12
Optic Neuropathy
Central nervous system (CNS) involvement occurs in up to 39% of patients with SLE.13,14 Optic nerve involvement is far less common and is estimated to occur in up to 1% of all patients with SLE.4,15
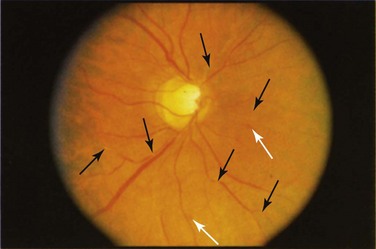
The pattern of optic nerve involvement varies in SLE. In some cases, patients present with symptoms and signs consistent with an acute optic neuritis,15,16 which is characterized by the acute onset of retrobulbar pain aggravated by ocular movement, an afferent pupillary nerve defect, visual field loss or scotomata, and either a normal-appearing or a swollen optic disc (Figure 31-7).
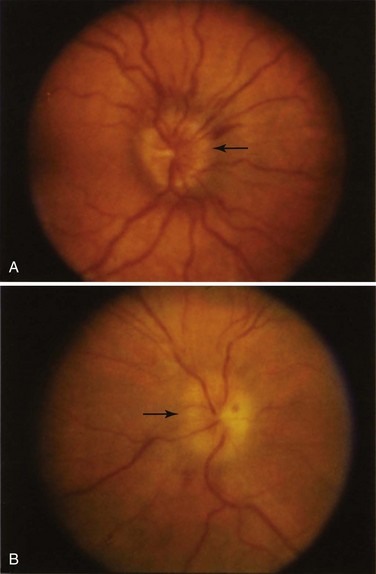
In other patients, the presentation may be more insidious such as a painless loss of vision that may be gradual in its onset17 with an afferent pupillary defect and an arcuate or altitudinal visual field defect evident on examination.
In both patterns of presentation, the pathogenesis is thought to be the same: microvascular occlusion leading to demyelination of the optic nerve in mild cases and optic nerve infarction in severe cases.15
The visual prognosis in individuals with SLE optic neuropathy is poor because it is notoriously difficult to treat. The standard treatment is corticosteroid therapy either orally or pulsed. However, recovery (if any) is slow, with final visual acuities of 20/200 or worse in several series.18
More recently, some success has been reported with the use of intravenous cyclophosphamide in addition to steroid treatment. Rosenbaum and colleagues16 reported a significant improvement in visual acuity and visual function in three patients treated with this regimen; a later series of 10 patients reported similar results with 50% of patients regaining normal visual acuity after treatment.19
SLE optic neuropathy is a rare cause of optic nerve disease, in comparison with other causes of optic neuritis, such as multiple sclerosis (MS). Certainly the distinction between SLE optic neuropathy with CNS involvement and MS can be difficult, because both may result in the same signs and symptoms and the two conditions have even been described to coexist.20 However, the response of SLE optic neuropathy to treatment is slow in comparison with the rapid response of MS-related optic neuritis.16
Episcleritis and Scleritis
Both scleritis and episcleritis may occur in patients with SLE21 and are a result of small vessel vasculitis affecting the tissues of the ocular coat—namely, the sclera and episclera.
Scleritis is a deeper and more severe inflammation than episcleritis and runs a more chronic course. It can result in visually debilitating complications if left untreated. These complications include severe scleral thinning (scleromalacia), resulting in the prolapse of intraocular structures, corneal scarring, glaucoma, and serous retinal detachments. Clinically, scleritis is often characterized by severe periocular pain, deep ocular injection, and significant tenderness of the affected area to palpation. Less commonly, its presentation may include severe pain and blurred vision without ocular injection if the posterior sclera is involved. More importantly, the development of scleritis is thought to be a serious development in SLE, because it has been described to parallel the degree of disease activity elsewhere.22–24
Corneal Disease and Keratitis
As previously mentioned, the most common ocular manifestation in SLE is KCS, which results in a poor tear film and secondary corneal changes such as small dry spots or epithelial erosions (e.g., superficial punctate keratopathy).1 However, other corneal disease may rarely occur, with a few cases of ulcerative keratitis and deep keratitis or inflammation of the deeper corneal layers (stroma) with secondary impairment of vision being described.25,26 These presentations are thought to be related to vasculitis affecting the surrounding limbal vessels. Ulcerative keratitis can also occur in association with scleritis.
Uveitis
Although described, uveitis, or intraocular inflammation, is an extremely rare association with SLE.22
Orbital Inflammation
Because SLE may affect any tissue of the body, orbital tissues such as the lacrimal gland (most commonly resulting in sicca), extraocular muscles, and other orbital tissues may also be involved, leading to symptoms of pain, proptosis, lid swelling, and diplopia. Such an orbitopathy from SLE alone is exceedingly rare, with only a handful of case studies being reported.21
Chloroquine and Hydroxychloroquine Toxicity
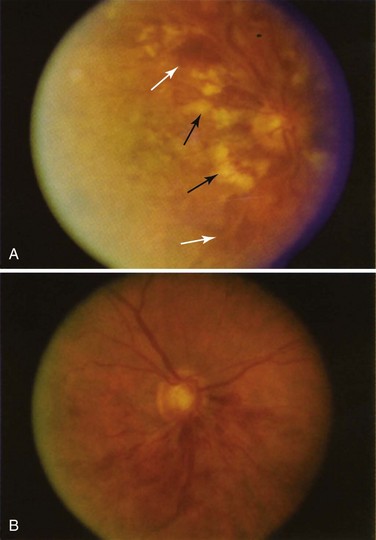
Chloroquine or hydroxychloroquine toxicity is classically characterized by the development of bilateral bull’s eye maculopathy that is visible on funduscopy (Figure 31-8). At this stage, observant patients may complain of a paracentral scotoma, whereas others may be asymptomatic. However, should drug exposure continue, further irreversible damage to the retina occurs that is discernible by retinal pigment atrophy, resulting in widespread retinal pigmentary changes and retinal vascular attenuation. By this stage, patients have severe visual field loss, decreased visual acuity, and impaired night vision.27,28
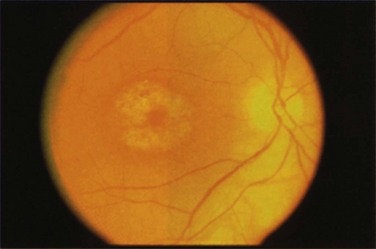
The first sign of toxicity is thought to be decreased visual function in the paracentral visual field that is detectable before the development of clinically visible bull’s eye maculopathy. Cessation of the drug at this early stage is thought to reverse these changes. However, once signs of maculopathy are clinically visible, these changes have been shown to be irreversible.28 In one recent series,29 progressive visual field loss or an electroretinographic abnormality was noted even after the medication was stopped.
The risk of retinal toxicity with chloroquine is higher than with hydroxychloroquine and occurs at lower doses (>3 mg/kg/day).28 A database study included nearly 4000 patients with either RA or SLE who had taken antimalarial medications. The study found that 6.5% of patients discontinued therapy because of ocular complaints. According to this study, weight and daily doses were not important factors in contributing to toxicity, but the duration of use was critical. After taking hydroxychloroquine for 5 years or longer, 1% of users had evidence of retinal toxicity.30 Several studies have found that ocular toxicity increases with doses greater than 6.5 mg/kg/day.29
In addition to dose and duration of therapy, risk factors have also been identified, such as co-existing retinal, renal, and liver disease, obese body habitus, and age older than 60 years.28 Research has also suggested that carriers of an ABCR-gene polymorphism, which has been linked to Stargardt disease, a form of hereditary maculopathy, may also be prone to developing either chloroquine or hydroxychloroquine toxicity at low doses despite a normal ophthalmic examination before treatment.31 Therefore the development of hydroxychloroquine (and chloroquine) toxicity may not be purely related to the dose but also to a variety of genetic and environmental factors.
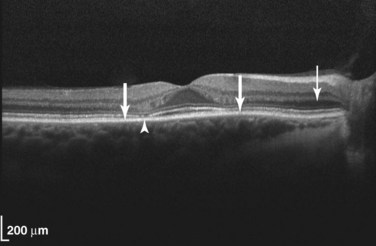
Several recommendations for regular ophthalmic screening of patients treated with chloroquine and hydroxychloroquine have been proposed to detect patients before they develop irreversible, severe vision loss. The American Academy of Ophthalmology currently recommends a baseline ophthalmic examination before commencing treatment. This examination should include a dilated fundal examination and the 10-2 Humphrey visual field test to assess the paracentral visual field.28 Three relatively recent screening methods have been cited by the American Academy of Ophthalmology for improved sensitivity for screening.32 These tests are (1) the multifocal electroretinogram, (2) the ocular coherence tomogram (OCT), and (3) autofluorescence imaging. One of these three types of studies should now be included in the assessment. Of the three techniques, OCT is the most standardized and widely available (Figure 31-9).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


