Chapter 39 Drug-Induced Lupus
Etiology, Pathogenesis, and Clinical Aspects
Etiology
Drugs Implicated
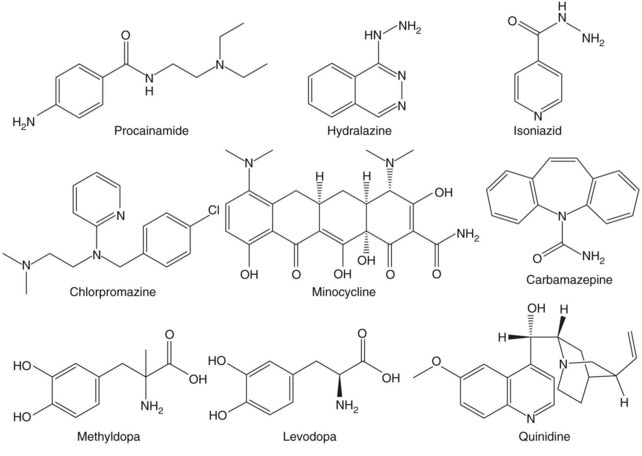
More than 80 drugs and recombinant therapeutic agents have been associated with lupus-like autoimmunity.1–3 For many, the evidence is based on case reports of lupus developing in patients receiving a drug and improving after its discontinuation. However, lupus is a chronic relapsing disease; therefore the relationship of drug ingestion to disease onset, flare, or remission may be coincidental, and some of the drugs reported to cause DIL may represent chance associations. Causality can be supported by documenting disease remission after the discontinuation of a drug and recurrence with repeat administration, which has been done for hydralazine,4 procainamide,5 isoniazid,6 chlorpromazine,7 quinidine,8 and minocycline.9 Prospective and case-control studies have provided additional confirmatory evidence for some of these and other drugs. Prospective studies support roles for procainamide,10 hydralazine,11 isoniazid,12 methyldopa,13 and levodopa,14 as well as estrogen and progesterone,15 in initiating lupus flares. Similarly, a matched case-control study of 875 participants with incident lupus in the United Kingdom General Practice Research Database reveals a significantly increased risk of lupus for patients receiving hydralazine, minocycline, and carbamazepine.16 Another case-control study confirms a significant lupus risk for estrogen/progesterone contraceptives17; this may be considered a form of DIL. These agents are listed in Table 39-1 together with their lupus risk or odds ratio, and the structures of the nonhormonal drugs are shown in Figure 39-1. Although methyldopa and levodopa have similar chemical structures and hydralazine and isoniazid share a hydrazine side chain, the other molecules have little in common, suggesting that no single chemical structure is responsible for inducing autoimmunity and that these drugs may induce autoimmunity by multiple mechanisms.
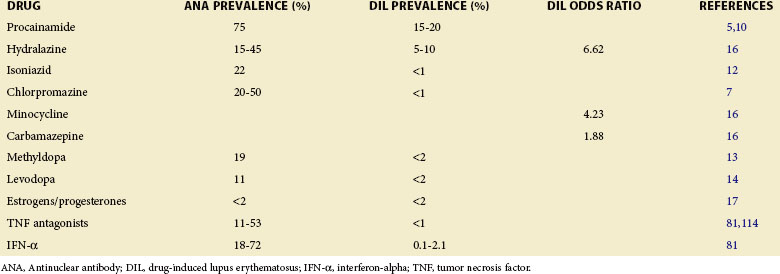
Nearly all of these drugs cause a positive antinuclear antibody (ANA) test more frequently than clinically overt DIL, and the duration of drug exposure required to develop DIL typically ranges from 1 to 3 years.10,11 Of these drugs, procainamide and hydralazine induce DIL most frequently with a 20% incidence after an average of 10 months of treatment with procainamide18 and 7% after 3 years with hydralazine.19 Oral contraceptives containing high doses of estrogens were also implicated in causing autoantibodies as well as clinical lupus in early reports. However, the more recent use of lower estrogen doses in combination pills may have decreased the incidence of DIL somewhat.20 Postmenopausal estrogen replacement has also been reported to cause lupus flares, but the flares are of mild to moderate severity.21 How estrogen influences autoimmunity is discussed in other sections of this textbook.
Finally, some recombinant biological agents, such as the tumor necrosis factor (TNF) antagonists and interferon-alpha (IFN-α), have been shown to cause lupus-like autoimmunity in prospective studies. These agents are also listed in Table 39-1. Like the lupus-inducing drugs, the TNF antagonists and IFN-α also cause ANAs more frequently than clinical lupus.2,22
Genetic Contributions to Drug-Induced and Idiopathic Lupus
Current evidence indicates that multiple genes are required for idiopathic lupus to develop. Those with a higher total genetic risk tend to develop lupus at an earlier age than those with a lower genetic risk, and those with a higher total genetic risk also develop anti–double stranded DNA (anti-dsDNA) antibodies and hematologic disorders (e.g., hemolytic anemia, lymphopenia, leukopenia, thrombocytopenia), as well as immunologic disorders (e.g., anti-Smith antibodies, antiphospholipid antibodies) more often than those with lower total genetic risk.23,24 This evidence suggests that people with even a lower total genetic risk are either unaffected or may develop ANAs only in response to environmental agents. Proposed environmental agents include infections, smoking, insecticides, silica, and ultraviolet (UV) light, among others.25–28
As with idiopathic lupus, drugs also activate lupus in genetically predisposed people, although the genetic associations with DIL are less well studied. Certain class II major histocompatibility complex (MHC) alleles predispose a person to idiopathic lupus,29 and a susceptibility to hydralazine-induced lupus has been associated with the human leukocyte antigen (HLA)–DR4 allele,30 although this observation was not confirmed by a second group.31 Unfortunately, the other genetic loci confirmed in idiopathic lupus are largely unstudied in DIL, and future studies could prove informative. Importantly, though, at least one genetic difference distinguishes DIL and idiopathic lupus. People who metabolize drugs slowly because of genetically determined slow acetylator status, perhaps as a result of N-acetyltransferases 1 (NAT1) or 2 (NAT2) polymorphisms,32 are more likely to develop DIL in response to drugs including hydralazine and procainamide than those who rapidly metabolize these drugs. In contrast, slow acetylator status does not predispose a person to idiopathic lupus.32
Age and Gender Contributions to Drug-Induced and Idiopathic Lupus
Age and gender also influence the susceptibility to DIL and idiopathic lupus. Although idiopathic lupus is usually considered a disease of young women, a recent study of more than 1600 patients with lupus in the British health care system demonstrates that the incidence of idiopathic lupus increases steadily up to approximately 75 years of age in men. In contrast, the incidence of lupus increases to approximately the age of menopause (50 to 54 years of age) in women but then declines, perhaps reflecting the decrease in estrogen after menopause, although some women still develop lupus well into their 80s.33
The age dependency in both men and women raises the possibility that environmental exposures have a cumulative effect in people genetically predisposed to lupus. As previously noted, those with a greater number of lupus genes develop lupus at an earlier age than those with fewer predisposing genes and have a more severe phenotype,23,24 supporting a gene-environment interaction in which a greater genetic predisposition permits lupus development with a lesser total cumulative environmental contribution; those with a lesser genetic predisposition require a more prolonged exposure. Interestingly, ANAs develop with age34 and often precede the development of clinical lupus by several years,35 also suggesting that cumulative effects of environmental exposures occurring over time in people with a lower genetic predisposition contribute to the development of ANAs and sometimes lupus later in life. The roles of age and the environment are discussed further in “Pathogenesis.”
In contrast to idiopathic lupus, drug exposure is required for DIL; consequently, the demographics of DIL differ from those of idiopathic lupus in part by reflecting the age of people receiving the drugs. With the exceptions of antiseizure medications more commonly used in younger people36 and minocycline used to treat acne in young women,37 older individuals tend to receive most of the drugs listed in Table 39-1. Perhaps, then, it is not surprising that DIL usually affects older adults more often than younger individuals.38 A case-control study reveals that patients developing minocycline-induced lupus were an average of 8.5 years younger than control participants developing idiopathic lupus.16 In contrast, the same study also found that people with hydralazine-induced lupus were approximately 25 years older at the time of disease diagnosis than patients with idiopathic lupus.16 Another factor potentially contributing to the increased incidence of DIL in older people is that drug metabolism slows with age,39 prolonging drug exposure in older adults, possibly analogous to slow acetylator status in procainamide- and hydralazine-induced lupus.40
Gender is also genetically determined, and women are affected by idiopathic lupus approximately nine times more often than men. Estrogen likely contributes to this female predisposition,20,21 as does having two X chromosomes.41 In contrast to idiopathic lupus, though, some forms of DIL occur only modestly more frequently in women.30,42 However, people receiving many of these drugs also tend to be older, and the decreased female-to-male ratio could also reflect declining estrogen levels in postmenopausal women.
Pathogenesis
The mechanisms by which most of the drugs listed in Table 39-1 cause autoimmunity are poorly understood. A number of possibilities have been considered, including drug metabolites acting as haptens to modify self-antigens and induce autoantibodies through epitope spreading; drug-induced cytotoxicity, releasing self-macromolecules; nonspecific lymphocyte activation; and a disruption of central tolerance in the thymus by the drugs themselves or by their reactive metabolites generated during inflammatory responses.1,43 However, although in vitro and in vivo models have been devised to support the feasibility for some of the mechanisms proposed, evidence that these mechanisms actually contribute to human DIL has been difficult to obtain for most of these drugs, and how the mechanisms might be relevant to idiopathic lupus remains unclear. Studies of procainamide and hydralazine, though, have provided important insights into epigenetic mechanisms contributing to both DIL and idiopathic lupus, whereas IFN-α and the TNF antagonists highlight important roles for these cytokines in the pathogenesis of idiopathic lupus. These mechanisms are discussed in the text that follows.
Epigenetics, Chromatin Structure, and Gene Expression
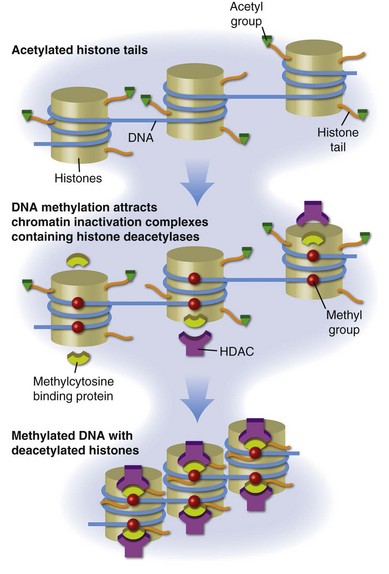
DNA is packaged into the nucleus as chromatin, the basic subunit of which is the nucleosome. Each nucleosome consists of two turns of DNA wrapped around a protein core containing two molecules each of histones H2a, H2b, H3, and H4. The nucleosomes are then arranged into higher order structures to form chromatin fibers. Histone tails protrude from the nucleosome, and positively charged lysines in the histone tails bind negatively charged phosphates in the DNA, stabilizing chromatin in a tightly compressed configuration (Figure 39-2). Chromatin in this configuration is inaccessible to transcription-factor binding and the messenger RNA (mRNA) transcription complexes responsible for gene expression. Gene expression thus first requires localized remodeling of the chromatin to make the DNA accessible, and these changes are replicated during cell division, making them heritable. The mechanisms regulating these changes in chromatin structure, which cause a heritable change in gene expression without a change in the DNA sequence, are referred to as epigenetic.44
Histone Modifications
One important epigenetic mechanism in chromatin remodeling is acetylation of lysines in the histone tails, referred to as histone acetylation. Acetylation of these lysines neutralizes their positive charge, releasing the histone tails from the negatively charged DNA and permitting localized remodeling of the chromatin into a transcription-permissive configuration open to transcription-factor binding (see Figure 39-2). Histone acetylation regulates gene expression in most if not all eukaryotic cells. Methylation, phosphorylation, ubiquitination, and other moieties also covalently modify the histone tail lysines to form a histone code that regulates other chromatin functions.45
Different cell lineages can express overlapping sets of transcription factors, potentially causing an expression of genes inappropriate for the function of different cell types. Histone modifications such as acetylation and deacetylation serve, in part, to facilitate appropriate gene responses and suppress inappropriate gene expression by permitting or preventing transcription-factor binding. However, this system is sensitive to the environment. Maintaining chromatin structure involves a dynamic balance in the activities of histone acetyltransferases (HATs), which add acetyls to histones, and histone deacetylases (HDACs), which remove them (see Figure 39-2), as well as the availability of acetyl groups, provided by acetyl-coenzyme A (acetyl-CoA). This process is potentially unstable because acetylation reactions are susceptible to environmental agents that affect the levels and activity of the HATs and HDACs, as well as intracellular acetyl-CoA levels, affecting gene expression. In higher eukaryotes and in particular vertebrates, chromatin is further stabilized in a condensed, transcriptionally silent configuration by DNA methylation.
DNA Methylation
DNA methylation refers to methylation of carbon 5 in cytosines to form deoxymethylcytosine (dmC). Methylated cytosines are found in mammals almost exclusively in cytosine-guanine (CpG) pairs. Methylcytosine-binding proteins such as MeCP2, MBD1, MBD2, and MBD3 bind dmC and tether HDAC-containing chromatin inactivation complexes to the methylated sequences, stabilizing the adjacent chromatin in a transcriptionally silent structure.46 The carbon-carbon bond between the methyl group and the deoxycytosine (dC) base is strong and resistant to enzymatic cleavage, providing a stable repressive mark on the DNA. The role of DNA methylation in silencing gene expression is also shown in Figure 39-2.
DNA methylation patterns are established during development by the de novo DNA methyltransferase 3a (Dnmt3a) and DNA methyltransferase 3b (Dnmt3b) and then replicated each time a cell divides by DNA methyltransferase 1 (Dnmt1), the maintenance methyltransferase. As cells enter mitosis, signals transmitted through the extracellular-regulated protein kinase (ERK) and jun-N-terminal kinase (JNK) pathways upregulate Dnmt1, which binds the replication fork in the dividing cells and reads CpG pairs. If a dC in the parent DNA strand is methylated, then Dnmt1 catalyzes transfer of the methyl group from S-adenosylmethionine (SAM) to the corresponding dC in the daughter strand, replicating the methylation pattern and producing SAM47 (Figure 39-3). This step is crucial, because if DNA methylation is inhibited, either by preventing Dnmt1 upregulation or by interfering with the transmethylation reaction, then the methylation patterns will not be replicated in the daughter cell. Consequently, genes that are normally silent can become demethylated and expressed. Further, since the changes are heritable, the errors will be replicated during subsequent cell divisions and will accumulate over time and, hence, with aging. Thus DNA methylation and, consequently, gene expression become sensitive to the environment at this point. Therefore drugs or chemicals that interfere with Dnmt1 levels or enzymatic activity and dietary or metabolic abnormalities that decrease SAM or increase S-adenosylhomocysteine (SAH) levels can inhibit DNA methylation, activating gene expression in a stable, heritable fashion; these errors will accumulate with age.48
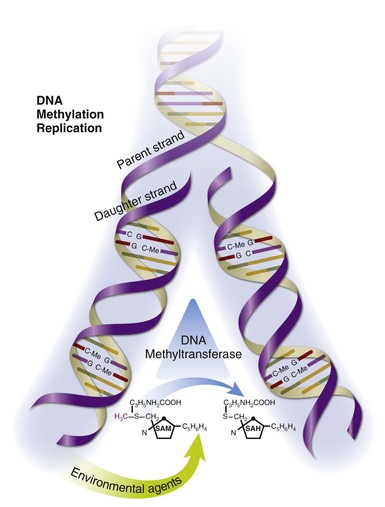
FIGURE 39-3 Replication of DNA-methylation patterns.
DNA-methylation patterns are replicated during mitosis by Dnmt1, which binds the replication fork and reads CpG pairs. If a deoxycytosine (dC) base in the parent strand is methylated (C-Me), then Dnmt1 catalyzes transfer of the methyl group from S-adenosylmethionine (SAM) to the corresponding dC in the daughter strand, replicating the methylation pattern and producing S-adenosylhomocysteine (SAH).47 This chemical reaction is susceptible to environmental agents such as procainamide, hydralazine, dietary abnormalities that affect SAM and SAH levels, and others.
Table 39-2 lists some of the recognized DNA demethylating agents and their mechanisms of action. These agents may affect gene expression in a wide variety of cells. However, T cells are particularly dependent on DNA methylation to regulate gene expression, and T cell DNA demethylation can contribute to lupus-like autoimmunity.
TABLE 39-2 Exogenous Agents and Proposed Mechanisms
| DRUG | MECHANISM | REFERENCES |
|---|---|---|
| Hydralazine | ERK pathway inhibitor | 67 |
| Procainamide | Dnmt1 inhibitor | 115 |
| Ultraviolet light | ERK pathway inhibitor | 72,116 |
| Aging | ERK/JNK pathway inhibitor | 116 |
| Diet | Modifies transmethylation reactions | 117 |
Dnmt1, DNA methyltransferase 1; ERK, extracellular-regulated protein kinase; JNK, jun-N-terminal kinase.
T Cells, DNA Methylation, and Drug-Induced Lupus
DNA methylation plays a critical role in regulating T lymphocyte effector and other functions. CD4+ T cells differentiate throughout life into multiple subsets, such as naive to memory, and T-helper 0 (Th0) to Th1, Th2, Th17, and T-regulator (Treg) cells. Although differentiation into these subsets is regulated by key transcription factors such as GATA-3 for Th2, T-bet for Th1, and FoxP3 for Treg, expression of many of the effector molecules, and therefore effector functions specific to these subsets, is regulated by a set of transcription factors that are expressed in more than one subset, potentially leading to inappropriate gene expression and cellular functions. Effector and other genes inappropriate for each subset are silenced by a repressive chromatin structure, stabilized by methylation of CpGs in their regulatory elements. Since DNA methylation patterns must be replicated each time T cells divide, interfering with DNA methylation during mitosis will induce expression of those genes normally suppressed by DNA methylation but for which the transcription factors are present. For example, treating dividing T cells with the irreversible Dnmt1 inhibitor 5-azacytidine (5-azaC) induces interferon-gamma (IFN-γ) in Th2 cells, interleukin 4 (IL-4) in Th1 cells, perforin in CD4+ T cells, FoxP3 in CD4+CD25− T cells, and killer-cell immunoglobulin-like receptors in CD4+ and CD8+ T cells.49,50
Inhibiting DNA methylation in CD4+ T cells also makes them autoreactive. CD4+ T cells normally recognize antigenic foreign peptides presented by self-class II MHC molecules on antigen-presenting cells (APC). Early studies demonstrate that DNA methylation inhibitors convert normal, antigen-specific CD4+ T cells into autoreactive cells that respond specifically to self class II MHC molecules on APC without the appropriate peptide in the antigen-binding cleft.51 The autoreactivity is caused by an overexpression of the adhesion molecule lymphocyte-function–associated antigen 1 (LFA-1) (CD11a/CD18). LFA-1 normally binds intercellular adhesion molecule 1 (ICAM-1) on APC and surrounds the T-cell receptor–MHC complex, forming the immunologic synapse. This binding stabilizes the T cell receptor–MHC interaction and provides additional co-stimulatory signals that result in T-cell activation when the T-cell receptor recognizes both the antigenic peptide and the MHC determinants. Treating CD4+ T cells with DNA methylation inhibitors, such as 5-azaC, increases LFA-1 expression. This allows the T cells to respond to the lower affinity interaction between the T-cell receptor and self-class II MHC molecules without the appropriate antigen, making the T cells autoreactive. Identical autoreactivity develops when T cells are transfected with LFA-1.52 Thus causing LFA-1 overexpression by treating with DNA methylation inhibitors or transfection is sufficient to break tolerance and cause MHC-specific T-cell autoreactivity.
Functionally, the autoreactive T cells recognize and respond to the self-class II MHC molecules on B cells, overstimulating antibody production through effects on both cell surface co-stimulatory molecules, as well as by secreting stimulatory cytokines similar to IFN-γ.53,54 The autoreactive T cells also respond to the self-class II MHC molecules on macrophages, but they kill them by inducing apoptosis, causing the release of apoptotic chromatin from the dying macrophages.55 Importantly, apoptotic chromatin is antigenic. Genetic deletion of any of the molecules involved in clearing apoptotic material causes lupus-like autoantibodies to nuclear antigens, including chromatin and DNA, as does overwhelming the clearance mechanisms by injecting apoptotic cells.56,57 Further, since macrophages are responsible for removing and degrading apoptotic chromatin, macrophage apoptosis will prevent clearance of the apoptotic debris left behind, augmenting autoantibody responses. The importance of macrophage apoptosis in autoimmunity was confirmed by experiments demonstrating that clodronate liposomes, which selectively deplete macrophages by apoptosis in vivo, cause lupus-like autoantibodies when injected into normal mice and accelerate nephritis in lupus-prone mice.58 Thus T cells made autoreactive by DNA methylation inhibition can generate both a source of antigenic chromatin by stimulating macrophage apoptosis, as well as overstimulating B cell responses to the nuclear antigens, contributing to the development of lupus-like autoimmunity.
The pathologic relevance of demethylated, autoreactive CD4+ T cells was first demonstrated in experiments during which murine CD4+ T cells were made autoreactive by treatment with the DNA methylation inhibitor 5-azaC and then injected into genetically identical mice. The modified cells caused anti-DNA antibodies and an immune complex glomerulonephritis.59 More recent studies used a transgenic mouse model to confirm that inducing T cell DNA demethylation in vivo causes lupus-like autoimmunity.60
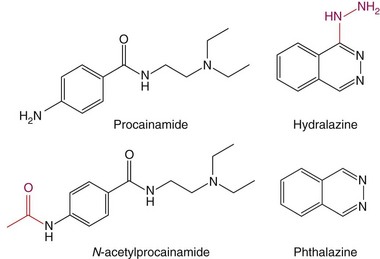
The observation that CD4+ T cells treated with a DNA methylation inhibitor cause lupus-like autoimmunity prompted studies testing whether drugs that cause lupus-like autoimmunity are DNA-methylation inhibitors. Procainamide and hydralazine, the two drugs causing DIL most often (see Table 39-1), were found to be DNA-methylation inhibitors, and treating CD4+ T cells with procainamide or hydralazine caused an LFA-1 overexpression and made CD4+ T cells autoreactive like 5-azaC. Procainamide was found to inhibit Dnmt1 enzymatic activity,61 whereas hydralazine was found to prevent Dnmt1 upregulation during mitosis by inhibiting ERK pathway signaling.62 Interestingly, N-acetylprocainamide, the acetylated metabolite of procainamide (Figure 39-4), does not cause DIL flares in patients with previous procainamide-induced lupus,63 is less potent in causing T-cell LFA-1 overexpression and autoreactivity in vitro, and has a reduced ability to cause autoimmunity in animal models.64 Other structure-function studies have compared hydralazine with phthalazine. Hydralazine is a phthalazine derivative with a hydrazine side chain (see Figure 39-4), and phthalazine is less potent than hydralazine in causing T-cell LFA-1 overexpression and autoreactivity and has reduced the ability to cause autoimmunity in animal models, suggesting that the hydrazine side chain may play a role.64
Related posts:
 The Innate Immune System in SLE
Clinical Aspects of the Antiphospholipid Syndrome
Gastrointestinal and Hepatic Manifestations
Novel Therapies for SLE: Biological Agents Available in Practice Today
Mixed Connective Tissue Disease and Undifferentiated Connective Tissue Disease
Pathogenetic Mechanisms in Lupus Nephritis
The Innate Immune System in SLE
Clinical Aspects of the Antiphospholipid Syndrome
Gastrointestinal and Hepatic Manifestations
Novel Therapies for SLE: Biological Agents Available in Practice Today
Mixed Connective Tissue Disease and Undifferentiated Connective Tissue Disease
Pathogenetic Mechanisms in Lupus Nephritis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


