Duchenne Muscular Dystrophy
DMD is an X-linked disorder with the chromosomal abnormality at the Xp21 gene locus (
7). As noted above, the gene codes for the protein dystrophin, an important cytoskeletal component of the muscle cell membrane. It appears that absence of dystrophin makes the muscle cell highly susceptible to mechanical stress, with eventual muscle fiber loss and replacement with fibrotic tissue (
5,
8).
DMD is the most common form of childhood muscular dystrophy, with an incidence of approximately 1:3,500 male births (
9). Although a male inheritance pattern is typical, as many as one third of cases may be due to new mutations, without any previous family history. Typical initial symptoms include abnormal gait, frequent falls, and difficulty climbing steps. Hypotonia and delayed motor milestones occur in earlier onset cases, but in 75% to 80% of cases, onset is noted before age 4 (
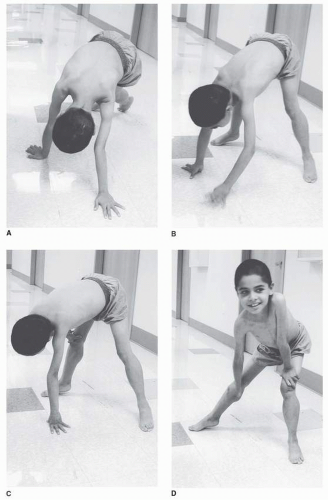
4). The abnormal gait is often noted by toe walking, which is a compensatory adaptation to knee extensor weakness, or increased lumbar lordosis as a compensation for hip extensor weakness. Another indication of pelvic girdle weakness is Gowers’ sign, demonstrated as the child rises from the floor. The patient generally begins by assuming a four-point stance, then brings the knees into extension while leaning the upper limbs forward, and sequentially moves the hands up the thighs until upright stance is achieved (
Fig. 30-2A-D).
On examination, the earliest weakness is seen in the neck flexors, typically during the preschool years. Weakness of the proximal musculature of the shoulder and pelvic girdle is next, with steady progression, although the patient and family may feel that functional loss does not occur gradually but rather quite suddenly. This may relate to a critical point in weakness or range of motion when compensatory actions can no longer suffice to perform a task. Quantitative strength testing shows greater than 40% to 50% loss of strength by age 6 years (
4), with fairly linear progression from ages 5 to 13 measured by manual muscle testing. Weakness appears to plateau after age 14 to 15, but this is probably a function of a floor effect and lack of sensitivity of the manual muscle testing scale (
10,
11).
Rehabilitation concerns are summarized in
Table 30-6. In patients not aggressively treated, the average age to wheelchair use is 10, with a range of 7 to 13 years of age. Prediction of transition to wheelchair use may be helped by using timed motor performance tests. In one natural history study, all DMD subjects who took more than 12 seconds to ambulate 30 ft lost the ability to ambulate within 1 year (
4). Immobilization, even for an acute illness, may lead to permanent loss of ambulatory ability during this phase of the disease.
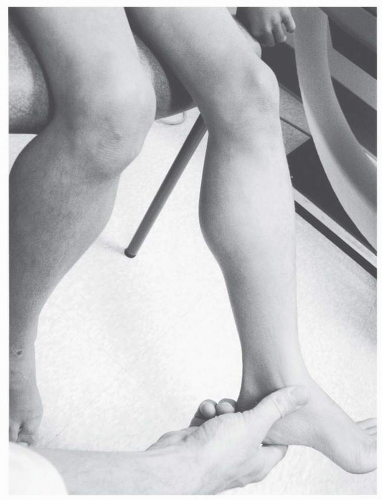
Unlike many myopathic disorders, joint contractures are a major concern in DMD. Nearly all affected boys older than 13 years have contractures (
4,
12,
13), and these contractures most commonly occur first in the ankle plantar flexors, iliotibial bands, and hip flexors, with subsequent involvement of the knee flexors and elbow and wrist flexors. There does not appear to be a strong correlation between less than antigravity strength for a muscle group and the severity of joint contracture, nor for strength imbalance between antagonists across a joint (
4). Clearly, lower extremity contractures become a problem after transition to a wheelchair for a significant part of the day. Natural history data suggest that progressive weakness, rather than heel-cord contractures, is associated with loss of ambulation as plantar-flexion contractures greater than 15 degrees are uncommon until after wheelchair reliance (
4) (
Fig. 30-3).
Scoliosis is a major clinical concern in DMD, and its prevalence is strongly related to age. Although significant curves often coincide with transition into wheelchair mobility, there does not appear to be a cause-and-effect relationship between scoliosis and wheelchair use (
4,
14). Rather, factors such as the adolescent growth spurt and progressive involvement of the trunk musculature may be responsible for progression of scoliosis during the adolescent years. There is some evidence that severity of scoliosis may be predicted by the type of curve and early pulmonary function measurements (
15). When the curves do not involve significant kyphosis or hyperlordosis and peak forced vital capacity (FVC) is greater than 2 L, severe progressive scoliosis appears less likely.
It is now clear that bracing does not slow the progression of spinal deformity (
12,
16,
17). Decision making for surgical management of scoliosis is closely related to pulmonary function. Although FVC volumes increase during the first decade of life close to 100% predicted with DMD, maximal static airway pressure (both maximal inspiratory and expiratory pressures) are impaired by 5 to 10 years of age. After a plateau in the early part of the second decade, there is progressive, fairly linear decline of FVC during adolescence (
4). A higher peak FVC obtained at age 10 to 12 may predict less severe restrictive lung disease and spinal deformity developing over the next few years (
4). An FVC below 40% predicted may contraindicate spinal instrumentation for scoliosis because of increased perioperative mortality; however, with current improved pulmonary care this is not an absolute contraindication (
18). Symptomatic respiratory failure in DMD typically manifests in later adolescence. Management of this complication is covered more in detail at a later section.
It is not surprising that cardiac function is affected in DMD, because the dystrophin protein has been shown to be present in both the myocardium and Purkinje fibers (
19). Most DMD patients older than age 13 demonstrate electrocardiogram (ECG) abnormalities (
4). The first abnormalities noted are Q-waves in the lateral leads, followed by elevated ST segments, poor R-wave progression, increased R/S ratio, and resting tachycardia and conduction defects (
4). ECG findings have been used to predict death from cardiomyopathy and include R wave in lead V, less than 0.6 mV; R wave in lead V
5 less than 1.1 mV; R wave in lead V6 less than 1.0 mV; abnormal T waves in leads II, III, aVF, V
5, and V
6; cardiac conduction defects; premature ventricular contractions; and sinus tachycardia (
20). Sudden death from ventricular ectopy, a complication of the cardiomyopathy and left ventricular dysfunction, is well described in DMD (
21,
22). However, progressive congestive heart failure is a more frequent sequela, and some investigators estimate that 40% to 50% of DMD patients die from this complication (
23,
24). Cardiomyopathy is usually noted after 10 years of age and occurs in nearly all patients by age 18 (
25). Cardiomyopathy is typically followed clinically with echocardiography, and the onset of systolic dysfunction is associated with a poor short-term prognosis (
26). Once patients with DMD reach adolescence, regular screening with ECG, echocardiography, and Holter monitoring is prudent.
Considering the presence of a dystrophin isoform in brain tissue (
27), it is not surprising that DMD patients show mildly decreased IQ scores compared with their peers and normative data (
4). There may be a specific deficit with tasks requiring attention to complex verbal information, regardless of IQ (
28). Mild impairments are noted on neuropsychological testing as well (
4), without a specific area of strength or weakness.
Obesity from reduced physical activity is a major concern in DMD, particularly at the onset of wheelchair dependence (
29,
30). Since many patients are now placed on corticosteroid treatment, weight gain is the most frequently reported side effect. At later stages of the disease (ages 17 to 21), significant weight loss becomes the predominant nutritional concern (
30,
31). This probably results from nutritional compromise along with increased protein and calorie requirements during the later stages of DMD (
32,
33), partially as a result of the increased work of breathing from restrictive lung disease.
At this time, there is no curative treatment available for DMD. Oral corticosteroids have been shown to increase muscle mass, increase strength, and slow muscle deterioration. However, the mechanism of its action is still unclear. Recent studies demonstrate additional potential benefits of corticosteroids including amelioration of cardiac, pulmonary, and scoliosis complications in DMD (
34,
35,
36). Research involving other pharmacoagents that can increase muscle bulk and strength as well as research into the stem cell and gene therapy are ongoing.
Becker Muscular Dystrophy
BMD is similar to DMD as an X-linked recessive disorder. It has a similar pattern of muscle weakness, but generally presents with a later onset and a slower rate of progression (
Table 30-7). Like DMD, the disorder has an abnormality in the gene location (Xp21) coding for the protein dystrophin. However, in this case, dystrophin levels are usually 20% to 80% of normal, or have the presence of the protein with an abnormal molecular weight. Mutation analysis of BMD has shown that majority are “in-frame” deletions, while DMD results from “frameshift” mutations. BMD is less common than DMD, with an overall prevalence recently estimated as 24 per million (
37).
Without dystrophin analysis, it may be difficult to clinically discriminate between DMD and BMD. Although age of onset typically occurs later in BMD, there is significant overlap with DMD (
38). The degree of CK elevation does not discriminate between the two diseases. The most useful clinical diagnostic discriminator is the ability to ambulate into adolescence. It is unusual for a patient with BMD to be wheelchair dependent before late adolescence, whereas even DMD outliers are dependent on the wheelchair for mobility by age 16. In fact, some BMD patients may be ambulatory well into middle age and beyond. There may be two distinct patterns of progression in BMD. In the first type, age of onset averages 7.7, and most patients have difficulty climbing stairs by age 20. In the more common milder form, age of onset averages 12, and there is no problem climbing stairs at age 20. The former group also seems to have a much higher rate of ECG abnormalities (
39). The percentage of normal dystrophin cannot be used to predict clinical course with any certainty in BMD (
40).
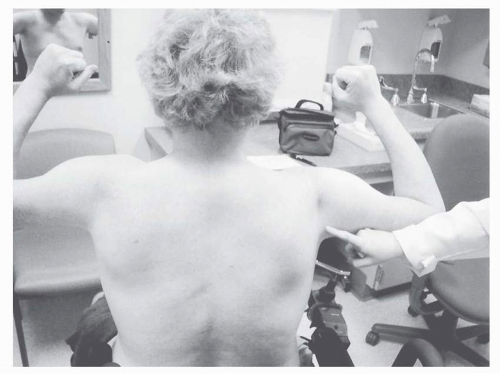
Findings on examination of the BMD patient mirror DMD, although milder. The neck flexors and proximal lower limb muscles are affected early, particularly the hip and knee extensors (
38). Subsequently, there is gradual involvement of the proximal upper-limb muscles (
Fig. 30-4). Extensors are
generally weaker than flexors (
38). Calf enlargement occurs, and presence of Gowers’ sign is indicative of the proximal muscle weakness. On standing, there is increased lumbar lordosis, and hip abductor weakness results in a waddling gait with trunk lean over the weight-bearing limb.
Contractures are not a significant early functional problem in BMD (
38,
39), becoming problematic only after wheelchair dependence. The joint locations for contractures are typical for one assuming the sitting posture, occurring in the hip flexors, knee flexors, and ankle plantar flexors. Significant scoliosis is much less common than DMD, and BMD patients rarely require spinal instrumentation (
38,
39).
One particular clinical concern in BMD is the potential for significant cardiac disease out of proportion to other manifestations of the myopathy (
39,
41,
42,
43,
44,
45). ECG abnormalities can be detected in about 75% of BMD patients (
38,
46). Most common abnormalities include abnormal Q-waves, right or left ventricular hypertrophy, right bundle branch block, and nonspecific T-wave changes. Echocardiography demonstrates left ventricular dilatation in 37% of BMD patients, and 63% have subnormal systolic function that is due to global cardiac hypokinesia (
46). Cardiac transplantation may even be necessary in some patients (
47,
48). The degree of cardiac compromise may not be reflected by clinical symptoms, and these patients should be screened at regular intervals with ECG and echocardiographic studies.
Unlike DMD, significant pulmonary dysfunction is not a hallmark of BMD. FVC does not fall below the predicted level until the third to fifth decade of life. Because of relatively greater involvement of the intercostals and abdominal musculature compared with the diaphragm, maximum expiratory pressure is compromised at an earlier age than maximal inspiratory pressure (MIP), similar to DMD (
38).
There are no consistent abnormalities on cognitive and neuropsychological testing in BMD other than a mild reduction in some patients (
38).