Chapter 46 Motor Neuron Diseases
On July 4, 1939, approximately 100 years after Sir Charles Bell first identified the motor function of the corticospinal tract and published pathologic findings from a middle-aged woman with limb paralysis, intact sensation, and anterior spinal cord degeneration, Lou Gehrig tearfully announced his retirement from baseball to a packed crowd in Yankee Stadium, proclaiming himself “the luckiest man in the world.”9,113 The United States watched as their beloved baseball hero nicknamed “the iron horse” suffered the ravaging effects of amyotrophic lateral sclerosis (ALS), the motor neuron disease (MND) with which his name remains synonymous. In 1941, at the age of 36, he succumbed to the disease, leaving his wife Eleanor to establish the ALS division of the Muscular Dystrophy Association in her pursuit of an illusive cure.
Classification of Motor Neuron Diseases
No universally accepted classification system exists for MNDs. Often the diseases are stratified into categories based on whether dysfunction is localized to UMNs, LMNs, or both. This strategy is complicated by the atypical MNDs such as progressive bulbar palsy (PBP), primary muscular atrophy (PMA), and progressive lateral sclerosis (PLS). These blur boundaries because their course often progresses from exclusively UMN or LMN involvement to frank ALS. Other strategies sort MND into categories based on pathologic process or group them under the umbrella terms typical and atypical motor neuron disease. Being familiar with the various organizational strategies is helpful when navigating the literature because it enables the reader to identify an author’s vocabulary bias and avoid unnecessary confusion (Box 46-1).
BOX 46-1 Motor Neuron Disorders
Upper and Lower Motor Neuron Disorders
Amyotrophic Lateral Sclerosis
ALS can be defined as a rapidly progressive neurodegenerative disease characterized by weakness, spasticity, and muscular atrophy with subsequent respiratory compromise leading to premature death. It is caused by the destruction of motor neurons in the primary motor cortex, brain stem, and spinal cord (Figure 46-1).8,113,139 ALS was identified as a clinical entity by Charcot in 1874 based on the gross histologic and clinical findings from 5 autopsies and 15 patient cases.113 “Amyotrophy” refers to muscular atrophy occurring from the degeneration of anterior horn cells in the spinal cord with muscle fiber denervation. “Lateral sclerosis” describes the resultant hardening of the anterior and lateral corticospinal tracts caused by replacement of dying motor neurons with subsequent gliosis.139
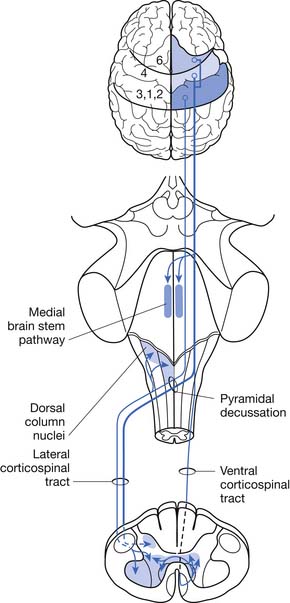
Onset of ALS is insidious and most commonly presents with painless asymmetric limb weakness. ALS most often afflicts people between 40 and 60 years of age with a mean age of onset of 58 years.100,106,109 Five percent of cases have onset before age 30.139 Men are affected more commonly than women with a ratio of 1.5:1.0. The worldwide prevalence is 5 to 7/100,000, making ALS one of the most common neuromuscular diseases in the world.40
Familial Amyotrophic Lateral Sclerosis
The vast majority of ALS cases are presumably acquired and occur sporadically. Approximately 5% to 10% of all ALS cases, however, are familial (FALS) and most commonly have an autosomal dominant inheritance pattern, although autosomal recessive, X-linked, and mitochondrial inheritance patterns have been reported.104,117,121 The age of onset of FALS occurs a decade earlier than sporadic cases, and progression of the disease is more rapid.80,93,134 Males and females are equally affected. About 20% of FALS cases result from a copper–zinc superoxide dismutase (SOD1) gene defect.9,110,134 These mutations are thought to cause disease by leading to a toxic gain of function from the abnormal tertiary structure, misfolding, of the protein rather than direct impairment in the antioxidant function of the SOD1 enzyme.8,119 More than 100 unique SOD1 mutations have been described.8,69,119,139 Other disease-causing genetic mutations have more recently been identified. These mutations have been found in genes encoding for angiogenin, chromatin-modifying protein, dynactin, vesicle-associated membrane protein, and TAR DNA-binding protein.139
Juvenile Amyotrophic Lateral Sclerosis
Juvenile-onset ALS by definition presents before age 25.139 It is a rarely occurring form of FALS. The progression of the disease is typically much slower than adult-onset ALS and can present initially with either UMN or LMN signs. Inevitably the disease progresses to encompass both UMNs and LMNs. Patients often maintain their ability to ambulate into midlife and can have a normal life span.
ALS2 is also inherited as an autosomal recessive trait. The disease-causing mutation has been linked to chromosome 2q33.41,139 Thus far, nine distinct mutations have been identified, all in the gene encoding for the protein alsin. Each mutation results in a premature stop codon causing production of a truncated, poorly functional protein product. Current research suggests that alsin is involved in vesicle transport and membrane trafficking.41 Disease onset typically begins before age 10. Prominent symptoms include limb and facial spasticity accompanied by pseudobulbar affect.
The autosomal dominant form of juvenile ALS, ALS4, presents with severe distal muscle weakness and pyramidal signs in the absence of bulbar and sensory abnormalities. It is caused by a mutation in the senataxin gene found on chromosome 9q34.41,139 The senataxin protein is known to have a role in RNA processing.41
Sporadic Amyotrophic Lateral Sclerosis
The etiology of sporadic ALS is unknown and likely multifactorial with a complex interplay of pathogenic cellular mechanisms.44,118 In a recent review by Wijesekera and Leigh139 published in 2009, they summarized nine cellular mechanisms for which there is mounting evidence to implicate their role in the pathogenesis of motor neuron degeneration. Included were genetic factors (already discussed), excitotoxicity resulting from excessive glutamate activity in the brain and spinal cord causing calcium influx into neurons with subsequent neuronal death, oxidative stress with accumulation of reactive oxygen species, mitochondrial dysfunction, impaired axonal transport, neurofilament aggregation, protein aggregation causing cytoplasmic inclusions, inflammatory dysfunction with abnormal microglial and dendritic cell activation, and deficits in neurotrophic factors with dysfunction of signaling pathways causing early cell apoptosis.
Population studies from the second half of the twentieth century suggested that the incidence of ALS was increasing. This was probably due, in large part, to increased life span and better recognition of the diagnosis.94,95 More recent population studies from multiple European countries have all demonstrated stable incidence.1,5,42,55,81 Many epidemiologic studies have been undertaken to find causal associations with minimal success. Consistent association between cigarette smoking and ALS, however, has been demonstrated.63 In a population-based case–control study conducted in three counties of western Washington state from 1990 to 1994, a twofold increase in risk was associated with a history of cigarette smoking, and a greater than threefold increased risk was observed for current smokers. Further, the authors found that alcohol consumption was not associated with increased risk of ALS; dietary fat intake was associated with an increased risk; and dietary fiber intake was associated with a decreased risk.96,97 Interestingly, consumption of antioxidant vitamins from diet or supplement sources did not alter the risk, but glutamate intake was associated with an increased risk of ALS.83,200,201 The finding that cigarette smoking and glutamate consumption increase risk for ALS appears consistent with current etiologic theories that implicate glutamate excitotoxicity and oxidative stress in the pathogenesis of ALS.
The hypothesis for an environmental cause of sporadic ALS has partially been spurred by evidence of considerable disease clustering demonstrated most profoundly in the Western Pacific region of the world.40,94,95 The prevalence in this region is 50 to 100 times higher than elsewhere. These populations include the Chamorro people of Guam and Marianas Island, the Kii Peninsula of Honshu Island, and the Auyu and Jakai people of southwest New Guinea, in whom ALS is associated with parkinsonism and dementia.139 Other sporadic cluster cases have been reported but without obvious environmental or causal factors.40
Clinical Features
Other signs and symptoms frequently associated with ALS are cachexia, fatigue, and musculoskeletal complaints. The term ALS cachexia refers to a phenomenon experienced by some patients in which weight loss occurs in excess of that caused by muscle atrophy and reduced caloric intake. Both subcutaneous fat and peritoneal fat are lost, presumably because of acceleration of the basal metabolic rate.116 In patients with ALS cachexia, more than 20% of body weight is typically lost over a 6-month period. Many patients with ALS feel an overwhelming sense of muscle fatigue, which is probably due to a combination of blocking of neuromuscular transmission in reinnervated nerve terminal sprouts and impairment of excitation contraction coupling.116 Some patients seek initial medical attention because of fractures or sprains that do not heal. In reality, these patients probably sustained their initial injury because of a fall or other injury (e.g., sprained ankle) that occurred because of underlying muscle weakness; they were then unable to recover to their premorbid level of function because of that weakness.
Poor prognostic factors include older age at time of onset, bulbar and/or pulmonary dysfunction early in the clinical course of the disease, short period from symptom onset to diagnosis, and predominance of LMN findings at diagnosis.40,100,107,109 More women than men present with bulbar symptoms, and the progression of bulbar palsy appears to be more rapid in women.96,97 Overall median survival from onset of symptoms in bulbar dominant cases is 2 to 3 years.39,73,100 In limb-onset ALS, it is 3 to 5 years.39,73,100 Young males with ALS can have a longer life expectancy, but overall the median 50% survival rate is 2.5 years after diagnosis. Survival rates will vary to a degree depending on the patient’s decision to use or not use mechanical ventilation and a feeding tube. Nonetheless, by 5 years postdiagnosis the overall survival rate is between 4% and 30%.39,40,100,109 Only 4% of patients will survive longer than 10 years.139
Atypical, “ALS-like” MNDs have been reported infrequently as a remote complication of several malignancies, including lymphoma, breast cancer, and small cell carcinoma of the lung.35,127 These likely represent paraneoplastic syndromes and not a true manifestation of ALS.111 Irrespective, patients with atypical MND should be screened for malignancy.
Selected Upper Motor Neuron Disorders
Progressive Lateral Sclerosis
PLS is a rare sporadic disorder of progressive spasticity with mostly spinal and occasionally bulbar region onset.64 Etiology of the disease is unknown. PLS can be clinically difficult to distinguish from hereditary spastic paraplegia (HSP) and is most reliably identified by the lack of familial inheritance.30 Onset of symptoms usually begins in the fifth decade; however, a juvenile variant also exists.30
Clinical Features
Spasticity is the most common presenting symptom. It is progressive and more often begins in the legs versus the arms or bulbar muscles. Asymmetric limb onset and spasticity involving the upper limbs or bulbar region are more common manifestations of PLS and can be diagnostically helpful in distinguishing PLS from the symmetric lower limb involvement observed in HSP.120 Limb wasting is rare in PLS and occurs in only 2% of patients.130 If disease onset is after the age of 45, prominent pseudobulbar symptoms with labile emotional affect can be problematic. Disease progression is usually slower than ALS, occurring over years to decades.120,130 Approximately 45% percent of patients diagnosed with PLS will eventually develop LMN symptoms and progress to ALS.64 For this reason, PLS is often considered a variant of ALS with early dominant UMN dysfunction as opposed to being a separate clinical entity. If the patient remains free of muscle wasting or other LMN symptoms for 4 years after diagnosis, he or she is less likely to progress to ALS, heralding better prognosis with greatly increased life expectancy.64,130
Hereditary Spastic Paraplegia
Like PLS, HSP is a disorder of UMNs and presents most commonly with progressive spasticity, weakness of the lower limbs, hypertonic urinary bladder, and impaired vibration sense. It is a genetically heterogenous group of neurodegenerative disorders in which the most severely affected neurons are those of the spinal cord.114 Caudal to rostral degeneration of the corticospinal tract and mild involvement of the dorsal columns occurs with progression of the disease.114 Population studies done in Ireland showed a prevalence of 1.27/100,000. Inheritance patterns include autosomal dominant, recessive, and X-linked forms. To date, 32 HSP loci and 11 HSP-related genes have been identified. They are classified as spastic gait locus 1 (SPG) through SPG33.57,114
Diagnosis is based on the clinical characteristics of the disease, neurologic examination demonstrating involvement of the corticospinal tract in both lower extremities, family history, and identification of a pathogenic mutation in a disease-causing gene. Sporadic cases occur providing the clinician with a diagnostic challenge. In a recent study by Brugman et al.,30 they determined that differentiation of sporadic presentations of HSP from PLS based solely on clinical characteristics was unreliable and therefore depends predominantly on genetic test results. Information on commercially available genetic tests and diagnostic laboratory locations can be found at http://www.genetests.org.30
Clinical Features
HSP is also classified by clinical characteristics and can be divided into “pure” or “complex” presentations. “Pure” HSP is considered the more classic pattern with spasticity, urinary disturbance, and vibration sense impairment.57,114 Onset occurs at any age, from early childhood through late adulthood, and progresses slowly over many years without exacerbations, remissions, or periods of abrupt worsening. Disability from spasticity and weakness is common, but life expectancy remains normal.57,114
“Complex” HSP is characterized by a combination of the above symptoms with concomitant neurologic disorders, such as seizures, impaired cognition, dementia, extrapyramidal disturbance, or peripheral neuropathy in the absence of other coexisting disorders such as diabetes mellitus, etc.57,114
Selected Lower Motor Neuron Disorders
Chronic Progressive
Progressive Muscular Atrophy
Progressive muscular atrophy by definition is a sporadic degenerative disease selectively affecting the anterior horn cells without signs of UMN involvement. Significant similarities between the natural history of progressive muscular atrophy and ALS have been observed, and debate continues as to whether progressive muscular atrophy should be considered its own clinical entity or a variant in the ALS disease spectrum.71,89 In a postmortem study of 12 patients thought to have progressive muscular atrophy, 50% of the autopsies demonstrated characteristic findings of ALS with degeneration of the corticospinal tract and ubiquitinated inclusions.71 Progressive muscular atrophy is a rare disease with unknown etiology.
Clinical Features
In a prospective study of 37 patients diagnosed with progressive muscular atrophy, published in 2007 by Visser et al.,136 the median age of disease onset was 57 years. The most common presenting symptom was distal limb weakness with muscle atrophy. The arms were affected slightly more often than the legs with either asymmetric or symmetric presentation. Bulbar symptoms were not typically evident at diagnosis but developed over the course of the disease in 43% of the patients. The onset of bulbar symptoms portended a more rapid decline with early death. During the 18 months of study, 35% of patients developed UMN signs progressing to an ALS phenotype. The 5-year survival rate was 45% with median survival of 56 months. Poor prognostic indicators included a forced vital capacity (FVC) less than 90% predicted at the time of diagnosis and decline in the FVC within the first 6 months after diagnosis.
Spinal Muscular Atrophy
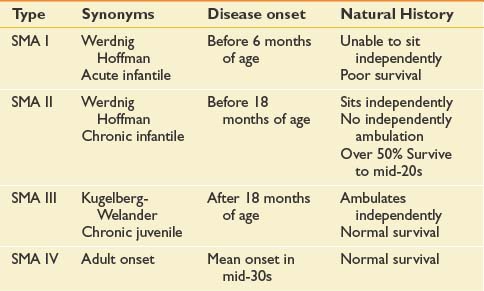
Many forms of spinal muscular atrophy (SMA) exist, all of which involve selective destruction of anterior horn cells. The various forms of SMA are clinically heterogenous, with some rare forms affecting distal or bulbar muscles only. The spinal muscular atrophies are mostly disorders with childhood onset and are usually inherited as autosomal recessive traits (Table 46-1). SMA is relatively common with an incidence of about 10 in 100,000 live births and a carrier frequency of 1 in 50.102
In 1990 the gene responsible for childhood-onset SMA was mapped to chromosome 5q11.2-13.3.31 Both the causative gene, named survival motor neuron 1 (SMN1, telomeric copy), along with a disease-modifying gene, survival motor neuron 2 (SMN2, centromeric copy) (32 to 35) were identified. The survival motor neuron (SMN) protein is ubiquitously expressed in all cells and tissues, with high levels in the nervous system, especially in the spinal cord.15 In about 95% of SMA patients, both copies of SMN1 exon 7 are absent or mutated. Recent data indicate that SMN1 deficiency alters stoichiometry of small nuclear ribonucleoproteins and leads to splicing defects for numerous genes in all cells, including motor neurons.141
Normal structural differences in the SMN2 gene cause frequent but not absolute exon 7 skipping during the splicing of SMN2 transcripts.24,140 The full-length transcripts of SMN1 and SMN2 encode proteins with an identical sequence. Without normal transcription of exon 7, the SMN protein product is less stable and more rapidly degraded, causing the neurodegenerative process. At least one copy of the SMN2 gene must be present in the setting of homozygous SMN1 mutations; otherwise, embryonic lethality occurs. The copy number of SMN2 varies in the population, and this variation appears to have important modifying effects on SMA disease severity.129 It appears an inverse trend exists between the number of SMN2 copies and phenotypic severity. Substantial variations in SMA phenotype and disease severity, however, can exist between patients with similar SMN2 copy numbers, suggesting other factors are likely involved. In the remaining 5% of SMA-affected patients, other small or subtle mutations have been identified.129,140
Clinical Features
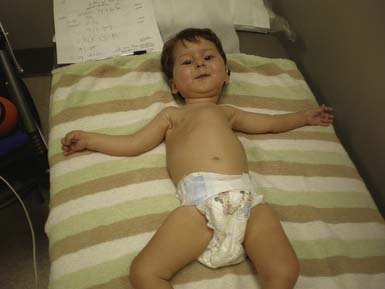
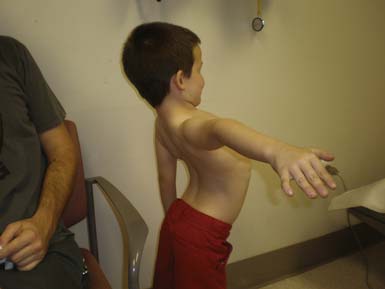
The most common forms of SMA are often referred to as types I, II, and III.32 SMA I, also known as Werdnig-Hoffman disease or acute, infantile-onset SMA, is a severe disorder often resulting in death before age 2 years, although recent studies have reported an increase in longevity likely resulting from better medical management of disease sequelae.83 Children with SMA I never attain the ability to sit independently (Figure 46-2). SMA II, also referred to as early-onset, intermediate SMA or chronic Werdnig-Hoffman disease, is less severe, with signs and symptoms becoming apparent in the first 6 to 18 months of life. These children will eventually attain the ability to sit independently but do not ambulate without assistance. SMA III, also known as Kugelberg-Welander disease, is a chronic, later-onset disorder, associated with significantly less morbidity. In SMA III, all early developmental milestones including the ability to ambulate independently are acquired (Figure 46-3). Signs and symptoms of SMA III usually become apparent between ages 5 to 15 years. Patients can be identified initially because of frequent falling and difficulty climbing stairs. In previous studies looking at SMA II and SMA III over a 10-year period, SMA II subjects showed marked weakness and progressive decline of strength, whereas SMA III subjects had a relatively static or very slowly progressive course and were far stronger. In both SMA II and SMA III, proximal weakness was greater than distal. Joint contractures, severe progressive scoliosis, and restrictive lung disease were present in most of the SMA II individuals, but these complications were less frequently identified in SMA III.32 Hand tremor, tongue fasciculations, and areflexia are common. Limb fasciculations occur more predominantly in SMA III. Intellectual function is preserved, and care should be taken to provide the child with SMA access to educationally stimulating environments.
SMA IV, also known as adult-onset SMA, has a mean age of onset in the mid-30s. It can be inherited as either an autosomal recessive or dominant trait.59,171 The disease clinically appears much like SMA III with slowly progressive proximal limb weakness and fasciculations. Deep tendon reflexes are either absent or depressed. Adult-onset SMA and SMA III patients can live normal life spans with a relatively benign disease course. Many of the rehabilitative modalities discussed in this chapter are applicable to this population. Further, with the rapid advancement of rehabilitation technology, many SMA II patients are now living well in to adulthood, and successful pregnancies have been reported in this population.33
Spinal and Bulbar Muscular Atrophy (Kennedy Disease)
Spinal bulbar muscular atrophy (SBMA) is often classified within the SMAs. It is, however, not associated with abnormalities in the SMN gene. It is a hereditary adult-onset disease that causes preferential degeneration of LMNs leading to weakness and atrophy of bulbar, facial, and limb muscles.12,58,128 SBMA is caused by a novel mutation, the expansion of a trinucleotide, cytosine–adenine–guanine (CAG) repeat, in the first exon of the androgen receptor gene and has X-linked recessive inheritance.12,24,58,128 Because CAG is transcribed to a glutamine, it is known as one of a few polyglutamine diseases. Unaffected individuals have a CAG repeat size that ranges between 5 and 35, whereas symptomatic individuals always have a repeat size of 40 or greater.24 Testosterone-driven accumulation of the mutated androgen receptor protein collects in nuclei and cytoplasm of motor neurons, resulting in their degeneration and loss.58
SBMA has some clinical variability; however, phenotypic expression does not appear to correlate with the length of CAG repeats. This is in contrast to myotonic muscular dystrophy and fragile X syndrome, where increased numbers of tandem triplet repeats correlate directly with disease severity.6
Clinical Features
Disease onset usually occurs after the age of 30 and is variable. Cases with onset in late adulthood have been reported. Patients present with amyotrophic, proximal, or distal weakness and wasting of the facial, bulbar, and limb muscles, sensory impairment, and endocrinologic disturbances, such as androgen resistance, gynecomastia, elevated testosterone or progesterone, and reduced fertility. Reflexes are typically decreased. There might be mild increases in creatine kinase. The course is slowly progressive with the ability to ambulate lost only late in life. Few patients require ventilatory support, and life expectancy is only slightly reduced.12,24,58,128
SBMA is clinically similar to ALS and often initially misdiagnosed. Degeneration of sensory neurons of the dorsal root ganglia is a common sign associated with SBMA, often preceding the onset of motor dysfunction in clear contrast to ALS. Electrodiagnostic studies demonstrating abnormal sensory nerve action potentials with spontaneous activity on electromyography (EMG) in patients with clinical signs similar to ALS should prompt further evaluation for SBMA. Another interesting difference to note is that Onuf’s nucleus, an androgen-sensitive spinal cord motor neuron nucleus that innervates the external sphincter muscles of the anus and urethra, is spared in ALS but degenerates in SBMA.6,45 Commercially available molecular genetic tests are now available for both SMA and SBMA.
Infectious
Poliomyelitis and Postpolio Syndrome
Acute poliomyelitis is a disease of the anterior horn neurons of the spinal cord and brain stem caused by poliovirus. The infection leads to the development of acute flaccid paralysis, which can be bulbar or spinal in distribution. The poliovirus is a small RNA virus belonging to the enterovirus group of the picornavirus family. Three antigenically distinct strains each have the potential to cause disease. Type I accounts for approximately 85% of cases progressing to paralytic illness. Only about 1% of patients who contract the disease will develop acute flaccid paralysis, also known as paralytic polio.74 Transmission occurs via fecal–oral route. The virus multiplies in the gastrointestinal tract during an incubation period lasting from days to weeks depending on the mode of infection (e.g., vaccine-induced vs. oral). The virus is shed in both saliva and feces during the incubation period. Further invasion of intestinal lymphoid tissues leads to hematologic spread and potential central nervous system involvement. The severity of the disease typically falls within a spectrum including no obvious manifestation of the disease, viremia without nervous system involvement, viremia with meningeal irritation but without paralysis, or paralytic disease with or without respiratory failure.74
Before the development of the polio vaccine, poliomyelitis was the most common cause of acute flaccid paralysis and disability in the United States. In the Northern Hemisphere epidemics were most common during the summer months. Peak incidence occurred during epidemics in the first half of the twentieth century, culminating in 1952 with approximately 58,000 cases reported in the United States.74 Children under age 15 were most commonly affected. Public fear and desperation led to the inception of the March of Dimes in the 1930s with a mission to eradicate the disease.84 The March of Dimes’ aggressive campaign raised public funds to support research and development of a vaccine. On April 12, 1955, Dr. Jonas Salk sparked celebrations around the world by announcing the results from the largest field trial in medical history.74 Salk had injected 325,000 second-graders with a trivalent inactivated polio vaccine, gave the same number of children a placebo, and monitored an even larger control group for disease. The vaccine nearly eliminated subsequent infection in the treated cohort and forged the path to conquer polio with initiation of the then-largest vaccination campaign in U.S. history. Since 1979 there have been no cases of wild-type poliovirus infection reported within the United States.
An oral vaccine developed by Albert Sabin from live attenuated poliovirus was introduced in 1963.74 Although the oral vaccine was less expensive to manufacture and simple to dispense, in January 2000, the Centers for Disease Control and Prevention (CDC) and the American Academy of Pediatrics recommended vaccination with the inactivated polio vaccine in an attempt to avoid further cases of paralytic polio in nonimmunized contacts of children receiving the live oral virus.7,38
In 1988, the World Health Organization, UNICEF, Rotary International, and the U.S. CDC launched the Global Polio Eradication Effort to rid the rest of the world of polio. Since the launch of this initiative, the worldwide incidence has declined. There remain four countries, however, where wild poliovirus is still endemic. Political instability in regions near the Pakistan/Afghanistan border and resistance of the population to polio immunization in northern Nigeria have resulted in insufficient vaccine coverage and remaining infections. In northern India low oral polio vaccine efficacy has thwarted efforts to eradicate the disease.43 In 2008, cases of poliomyelitis were reported in countries where none occurred in 2007. These countries included Sudan, Benin, Ethiopia, Burkina Faso, and Nepal, illustrating the importance of continued widespread vaccination prevention programs.50
Clinical Features
In patients with overt manifestations of infection, symptoms consist of fever, malaise, myalgia, sore throat, and gastrointestinal upset. Aseptic meningitis with headache, back pain, and stiff neck develop with increasing severity of the disease. In the approximately 1% of patients who progress to paralytic disease, localized fasciculations with intensely painful myalgias occur after 2 to 5 days of illness.74 Asymmetric weakness and atrophy affecting the legs more often than either the arms or bulbar muscles progress to flaccid paralysis. Dysautonomia including labile blood pressure, cardiac arrhythmia, and gastrointestinal and urinary dysfunction can require emergent medical intervention and is associated with higher rates of mortality.74 Respiratory failure often develops rapidly if infection involves the medullary respiratory center or causes weakness of respiratory muscles. Paralysis remains static for several days to weeks followed by slow recovery over months to years. Improvement of strength after acute paralytic polio occurs both by recovery of some neurons and sprouting from remaining axons innervating locallydenervated muscle fibers.84 The enlarged motor units can be up to 8 times the normal size. Recovery is often incomplete with residual weakness and disability.84
Late effects of poliomyelitis occur more commonly in patients with history of paralytic polio. Farbu et al.56 prospectively examined 85 patients with late effects of polio and found that the most common complaints were pain, muscular weakness, and fatigue. They identified loss of function resulting from degenerative joint disease with and without nerve entrapment in 53% of patients. Compensatory mobility patterns secondary to limb weakness and side-to-side growth disparity, as well as overuse of unaffected limbs, have been blamed for increased incidence of symptomatic osteoarthritis.
Of the 85 patients studied, only 26% met criteria for the diagnosis of postpolio syndrome (PPS; Box 46-2). Gradual or sudden onset of new progressive weakness and decreased muscular endurance after a period of at least 15 years of functional stability suggest PPS. The etiology of PPS is unknown. Proposed mechanisms for the development of PPS include distal degeneration of surviving enlarged neurons resulting from increased metabolic demand, degeneration of terminal axonal sprouts resulting in muscle fiber denervation, neuromuscular junction dysfunction as demonstrated by increased jitter on single fiber EMG, and loss of neurons through the normal aging process.56,74,84 Careful medical evaluation should be undertaken to exclude other possible and potentially treatable causes of the patient’s complaints.
BOX 46-2 Criteria for Diagnosis of Postpolio Syndrome
Modified from the March of Dimes: International conference on post-polio syndrome identifying best practices in diagnosis and care, May 19-20, 2000. Available at: http://www.marchofdimes.com/files/PPSreport.pdf.
Clinical Evaluation in Motor Neuron Diseases
History
The evaluation of a patient suspected of having MND begins with a detailed history and general physical and neurologic examination. The history should establish the age at the time of onset and the initial presenting symptoms. The clinician should identify the pattern of weakness and/or spasticity. Are symptoms symmetric versus asymmetric, distal versus proximal, limb or bulbar predominant? Asking the patient what activities have become more difficult can give clues to the pattern of weakness. For example, a patient with proximal weakness might complain of difficulty brushinghis or her hair or climbing stairs. It is also important to determine the rate and pattern of progression of symptoms because these can give diagnostic and prognostic clues. A detailed past medical, social, and family history should be obtained, exploring the potential of immune-mediated, toxic, infectious, or familial etiology.
Physical Examination
On neurologic examination, one is looking for evidence of UMN and LMN dysfunction (Box 46-3). The mental status, nonmotor cranial nerve function, sensory examination, and cerebellar examinations should be normal in the patient with ALS but can be abnormal in atypical MND. Findings of upper motor involvement on examination include spasticity and hyperreflexia, indicated by abnormal spread and amplitude of reflexes, clonus, or by the presence of reflexes despite muscle atrophy as a result of LMN loss. The gold standard used to diagnose UMN pathology is the presence of pathologic reflexes, such as the Babinski sign, Hoffman sign, jaw jerk, and palmomental and snout reflexes.28 If the toe extensors are paralyzed, visualization of contraction of the tensor fascia lata when an attempt is made to elicit a Babinski response has the same significance as great toe extension. Recently, it has been suggested that the corneomandibular reflex might be a more sensitive and specific indicator than the jaw jerk of UMN pathology in the bulbar region.10
LMN findings on examination include weakness, atrophy, hypotonia, hyporeflexia, and fasciculations. Head drop is a manifestation of muscle weakness often seen in ALS, although it can be seen in other neuromuscular disorders. ALS and myasthenia gravis are the two most common causes of head drop (Figure 46-4). Fasciculations are common in lower MND and can occur in the tongue and the extremities.

Electrodiagnostic Testing
No biomarkers are currently available for the diagnosis of ALS. Diagnosis is based on clinical findings requiring the presence of signs of UMN and LMN degeneration and the progression of the symptoms from a body region to another. Electrodiagnostic testing is considered an extension of the physical examination and should be performed on patients suspected of having an MND. Purely UMN disorders will have normal electrodiagnostic studies. The various forms of MND with involvement of the LMN share several electrodiagnostic features. General electrodiagnostic testing characteristics of MND include normal sensory nerve conduction studies with the exception of Kennedy disease, normal or low motor amplitudes depending on disease stage, and normal distal motor latencies and conduction velocities. With profound loss of motor amplitude, conduction velocities can decrease as low as 25% below the lower limit of normal because of loss of the fastest conducting fibers. Motor nerve conduction studies, including proximal stimulation sites to assess for conduction block resulting from peripheral neuropathy (e.g., multifocal motor neuropathy with conduction block) should be included in the electrodiagnostic plan.48
The needle electrode examination shows a decreased recruitment pattern, either normal size or large motor unit action potentials with or without evidence of remodeling depending on the specific disease process, and abnormal spontaneous activity including positive sharp waves, fibrillation potentials, fasciculations, and complex repetitive discharges.48 The electrodiagnostic study should include needle examination of the thoracic paraspinal muscles.49 Choosing to sample muscles below the T6 root level helps to avoid confounding results caused by multilevel root innervation from the lower cervical segments. Other muscles that can be sampled that are perhaps unique to MND include bulbar, facial, masticatory, and rectus abdominus muscles. These can be useful to find evidence of involvement in additional body regions needed to meet the El Escorial criteria for the diagnosis of ALS.48
Other electrodiagnostic tests to consider in the setting of an MND evaluation include single-fiber EMG, which might demonstrate increased jitter resulting from immature sprouting of nerve terminals during reinnervation in ALS. Motor unit number estimation assesses the size of a motor unit and can be followed over time for changes from motor neuron loss and subsequent sprouting of remaining neurons leading to enlarged motor units. Motor unit number estimation is being evaluated as a potential outcome measure in ALS clinical trials.48 Transcranial magnetic stimulation holds promise in evaluation of UMN dysfunction; however, it is not widely available, and this limits its clinical use.48
El Escorial Criteria
The El Escorial Criteria for diagnosing ALS were developed by a task force of the World Federation of Neurology in 1990 to ensure inclusion of more homogeneous patient populations in ALS clinical trials.27 These criteria have been used to enroll patients in most of the recent clinical trials. The criteria were revised in 1998 and again at the international symposium held in Awaji-shima, Japan, in December 2006 with the intent to improve the speed and certainty of diagnosis.27,48 The current criteria classify the certainty level of the diagnosis of ALS as falling into one of three categories: definite, probable, and possible. The category of “Laboratory Supported Probable ALS” is no longer included based on the consensus panel’s decision that clinical features of neurogenic change and neurogenic EMG findings should have the same diagnostic significance in an individual muscle and can be considered together in a single limb to meet the required abnormalities for diagnosis of ALS.48
The El Escorial criteria divide the motor system into four regions: bulbar, cervical, thoracic, and lumbosacral. Clinical evidence of UMN and LMN pathology is sought within each region. The certainty level of diagnosis depends on how many regions reveal UMN and/or LMN pathology. Box 46-4 summarizes the schema for placing patients in the three diagnostic categories.
BOX 46-4 EI Escorial Criteria
Modified from the Awaji-shima Consensus Recommendations.48ALS, Amyotrophic lateral sclerosis; LMN, lower motor neuron; UMN, upper motor neuron.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


