Chapter 54 Myelomeningocele and Other Spinal Dysraphisms
Myelomeningocele (MMC) is the most common congenital anomaly of the central nervous system. It exists in a spectrum of neural tube defects (NTDs), ranging from cranioschisis, or complete failure of neurulation, at one end to spina bifida occulta, with minimal or no neurologic involvement, on the other end.92 Although the most severe NTDs result in stillbirth or death shortly after birth, the majority of patients with a spinal dysraphism survive. MMC, which composes 90% of open spinal dysraphic states, is the most complex congenital anomaly compatible with life and is the second most common disabling condition in childhood after cerebral palsy.20,119,169 The varying degrees of organ involvement seen with these conditions have major implications for long-term health and physical function, as well as the psychologic and social well-being of the affected individual. Survival and quality of life for those with MMC have improved because of advances in medical, surgical, and rehabilitative care during the past 50 years. Many challenges remain, however, regarding advancing prevention, understanding etiology, maximizing health-related outcomes, transitioning to adult-based health care systems, and improving activity and participation at the societal level. These challenges are best met by a team of specialists working with patients, their families, and the community.
Terminology and Historical Background
The first clear description of spinal dysraphism was by Casper Bauhin, a Swiss physician, anatomist, and botanist, in his publication Theatrum Anatomicum in 1592. The term spina bifida, however, is often historically associated with Nicholas Tulp, a Dutch physician who published a sketch (Figure 54-1) and description of several patients with the condition in 1641. In 1875, Rudolf Virchow described spina bifida occulta, which refers to a hidden bony defect, as well as other potential hidden anomalies.165 Spina bifida has been further delineated with the term spina bifida aperta, a midline defect that communicates with the external environment and includes MMC and meningocele. The term spina bifida cystica is also sometimes used, and simply refers to a sac filled with cerebrospinal fluid protruding from the spinal column but can also refer to MMC and meningocele. MMC at the level of the spine refers to protrusion of the meninges through a defect in the posterior elements of the spine, with involvement of the spinal cord or nerve roots. Meningocele refers only to protrusion of the meninges and cerebrospinal fluid through a defect in the posterior elements of the spine into the tissue beneath the skin, without involvement of functional neural elements. These distinctions are important and will be further explained below.
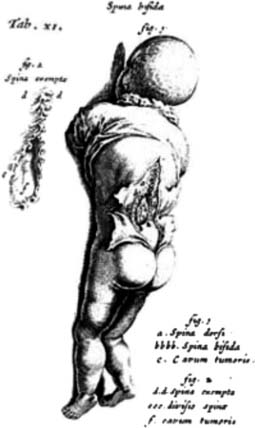
Because this terminology is often confusing and minimally descriptive, an alternative system based on advances in clinical recognition and imaging of spinal lesions now allows for more useful classification, especially as it relates to prognosis. This point is exemplified by studies reporting a 17% incidence of bony spina bifida occulta in the general population and in 30% of normal individuals aged 1 to 10 years with no neurologic involvement or anatomic abnormality other than incomplete closure of the posterior elements of the spine.16,91 In contrast, occult spinal dysraphism, including diastematomyelia, lipomyelomeningocele, and tight filum terminale, among others, can present with neurologic compromise or orthopedic deformity, with no further outward anatomic abnormalities other than possible cutaneous anomalies. Modern imaging studies are able to reveal these underlying defects and provide a system of classification based on neuroradiologic and clinical findings.178
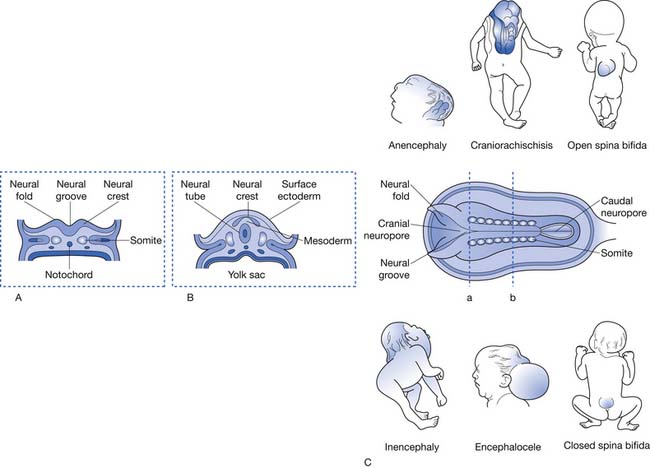
This system of classification simply divides the various forms of spinal dysraphism into open spinal dysraphisms (i.e., open spina bifida) and closed spinal dysraphisms (i.e., closed spina bifida) (Figure 54-2). It is useful from a prognostication standpoint because it is based on the observation that those with open defects generally have lesions with visible neural elements, often leaking cerebrospinal fluid, which are associated with malformations that involve the entire central nervous system, including Chiari II malformations, midline defects, and hydrocephalus. In contrast, closed spinal dysraphisms (meningocele and occult spinal dysraphism) are lesions that are fully epithelialized with no neural tissue exposed, and generally have the malformation limited to the spine and spinal cord, with only rare involvement of the brain.3 Box 54-1 displays a complete listing of spinal dysraphic states.178 Although most of the conditions listed in Box 54-1 can result in a patient having rehabilitation needs, the most severely affected and vast majority of affected individuals will have a history of MMC. For this reason, the remainder of this chapter refers to rehabilitation concepts as they apply to MMC, although they can be applied to the various other spinal dysraphisms as appropriate.
BOX 54-1 Cliniconeuroradiologic Classification of Spinal Dysraphism
From Tortori-Donati P, Rossi A, Cama A: Spinal dysraphism: a review of neuroradiological features with embryological correlations and proposal for a new classification, Neuroradiology 42(7):471–491, 2000, with permission.
Epidemiology
Incidence and Prevalence
Epidemiologic studies of spinal dysraphisms typically include both MMC and meningocele, which are collectively referred to as spina bifida.18 One clear observation from these studies is that the prevalence of MMC has decreased worldwide during the past century.3 In the United States, rates have varied from as high as 2.31/1000 births in Boston during the 1930s to as low as 0.51/1000 births in Atlanta in the early 1990s.127,144 The most recent U.S. data indicate a 6.9% decrease in prevalence, from 2.04/10,000 live births from 1999 to 2000, to 1.90/10,000 live births from 2003 to 2005.34 Rates in other parts of the world have been as high as 3.4/1000 live births in Dublin, Ireland in 1953, although these rates have generally decreased in a fashion similar to that in the United States.38
The reasons for the decreasing prevalence of MMC are multifactorial and not completely understood. A portion of the decrease can be attributed to the advent of prenatal screening and elective termination of pregnancy.42,114 This is only partially responsible, however, and other factors are known to play a role. The most influential factor has been the increased consumption of folic acid among women of childbearing age. Folate supplementation was first shown to decrease rates of NTDs in studies performed in Wales in the early 1980s.93 Multiple observational and controlled trials followed, leading up to the Medical Research Council study, which involved seven countries and 3012 women. The study was ended early after supplementation with folic acid (4 mg/day) was shown to prevent 72% of NTDs in women with a previously affected pregnancy.115 A subsequent study proved the efficacy of folic acid in preventing the first occurrence of an NTD.43 Both recommendations by the U.S. Public Health Service beginning in 1992 for daily folic acid intake for women of childbearing age and mandatory supplementation of all enriched cereal grain products in the United States since 1998, however, have not resulted in the 48% reduction in NTDs predicted by these studies.44 A decrease of approximately 26% has been observed, illustrating the need to further educate all women of childbearing age on the importance of daily folic acid intake of 0.4 mg for those who could become pregnant and 4 mg for those with a previously affected pregnancy or a history of MMC themselves. These statistics also place emphasis on the importance of health care provider participation in this effort.35 A recent study showed that women aged 18 to 24, who account for almost one third of U.S. births, had the least awareness regarding the need for folic acid consumption, appropriate timing of consumption, and the lowest daily use of supplements.188 Also of note, folic acid supplementation was ineffective in decreasing the overall incidence of lipomyelomeningocele between 1995 and 2001 in Nova Scotia in one study, providing evidence that the embryogenesis of various spinal dysraphisms could be fundamentally different.114 Subsequent studies have shown a more substantial reduction in cranial, cervical, and thoracic defects related to folic acid consumption compared with lumbar and sacral defects, further pointing to the etiologic heterogeneity of NTDs.46 A recent U.S. study conducted after the implementation of fortification of the food supply found little evidence of a link between maternal folic acid intake and NTDs. The authors theorize that folic acid fortification has reduced the folic acid–sensitive NTDs, with those that remain resulting from additional risk factors or underlying mechanisms.121
Geographic variation in birth prevalence of NTDs occurs both between and within countries.86 Across Europe, rates have been noted to be higher in Germany and Hungary than in Scandinavian countries. In the United States, there has been an observed trend of decreasing rates of NTD from the east coast to the west coast.64 Ethnicity has been shown to play a role in MMC as well, with those of Celtic descent (Irish, Welsh, and Scottish) having higher rates than those of Anglo-Saxon or Norman origin. These findings have been correlated with the high rates of MMC in Boston in the 1930s, which were highest among those who had mothers of Irish descent.30,128 In addition, those of Hispanic, Chinese, and Sikh ethnicity are at a greater risk for MMC.34 (1) Rates among black and Asian populations have been observed to be low, with similar rates among populations living in different areas.86 Female gender has also consistently been shown to be a risk factor for MMC, with a female preponderance observed in both still and live births.84,86 Additionally, the prevalence of spina bifida in Texas along the Mexico border was recently reported to be 3.52/10,000 live births, well above the most recent Centers for Disease Control and Prevention data for the entire country.28
Embryology and Etiology
Embryology
NTDs are known to occur as a result of failure of neurulation between the seventeenth and thirtieth days of gestation.74 Primary neurulation refers to the development of the neural tube, which forms the brain and spinal cord. Secondary neurulation refers to formation of the remainder of the neural tube from a cell mass caudal to the posterior neuropore, which forms the lower sacral and coccygeal segments. The caudal neuropore closes around the twenty-sixth day of gestation, and, as a result, teratogenic events that take place after this closure cannot cause thoracic or lumbosacral MMC.94,161 Failure of primary neurulation can lead to an open NTD, consisting not only of a spinal anomaly but also other defects, including a Chiari II malformation and hydrocephalus, possibly resulting from cerebrospinal fluid loss during early development.82,112 Most posterior lumbar and sacral meningoceles are thought to occur during secondary neurulation, with those higher on the spinal axis resulting from defects in primary neurulation that do not cause an open NTD.3
Etiology
Although the mechanisms are not well understood, up to 80% of all NTDs are thought to be due to multifactorial influences (i.e., genetic and environmental factors). Certain other NTDs are associated with NTD syndromes, single-gene disorders, and chromosomal disorders and are relatively well defined.5 Recurrence rates of NTDs are influenced by family history, geography, and severity of anomaly. These rates have been reported to vary from 2.4% to 5% after the birth of one affected child, with the risk doubling after two affected children.40 A Hungarian study reported that recurrence risk among those with spina bifida was greatest for MMC associated with hydrocephalus, at 4.79%.137 In addition, studies in the early 1990s showed that at least 70% of NTDs are “folic acid–sensitive” or “folic acid–dependent,” with the remaining 30% being “folic acid–resistant.”6 Recent studies from Ireland have confirmed that both homozygosity and heterozygosity for the T allele of the C677T polymorphism of the gene encoding the folate-dependent enzyme 5,10-MTHFR are risk factors for NTDs. This polymorphism is associated with lower tissue folate concentrations, higher homocysteine concentrations, and lower enzyme activity than in the wild-type genotype. This single genetic variant could account for up to half of the folate-related NTDs in Ireland.88 These combined at-risk phenotypes are present in approximately 59% of the European population and 53% of the North American population.19 Recent studies have also investigated links between genetic dysregulation of platelet-derived growth factor, myoinositol, and nitric oxide synthase, among others, as contributors to the formation of NTDs.25,66,207 Two large trials are currently ongoing to further elucidate the genetics of spina bifida and other NTDs.118
A number of environmental factors have been implicated in the development of NTDs, including low socioeconomic status130,196; maternal diabetes mellitus190; maternal hyperthermia153; folate deficiency117; hyperzincemia113; maternal obesity195; and certain drug exposures, including to carbamazepine,146 valproic acid,135 diuretics, antihistamines, and sulfonamides.125 Of the medications mentioned, carbamazepine and valproic acid have the strongest correlation with spina bifida, with an estimated risk of 1% and 1% to 2%, respectively.135,146 Seasonal variation, with peaks in midspring conception for spina bifida, has been reported in some regions but has not been confirmed in others.33,60 The factors for which there is more convincing evidence of an association to NTDs, however, include maternal obesity, maternal hyperthermia, and lack of maternal periconceptional folic acid supplementation.118 As well, a recent Canadian study suggests that vitamin B12 supplementation in combination might be more effective in preventing NTDs than folic acid alone.177
Prenatal Counseling (Diagnosis and Management)
Prenatal screening now allows for diagnosis of the majority of cases of MMC before birth. Initial screening of women who are at high risk, including those with a positive family history, a previous child with spina bifida, or exposure to teratogenic agents, should include measurement of serum α-fetoprotein and acetylcholinesterase levels at 16 to 18 weeks postconception. Based on these results, a patient-specific risk can be calculated and repeat testing of serum levels performed. Subsequently, high-resolution ultrasound can be performed, which is sensitive in 95% of cases in which good images are retrieved. If the images are of poor quality because of maternal obesity or other factors, amniocentesis can be performed to obtain amniotic fluid α-fetoprotein and acetylcholinesterase levels. These tests can be repeated based on previous equivocal results; however, after diagnosis, genetic counseling along with a discussion of management options should occur. A recent study showed the diversity of physician views regarding prenatal screening, selective termination, and disability, suggesting a need to better understand how these differences affect reproductive technology, health care policy, and medical practice.199
Prenatal detection of MMC is important to educate the patient and family regarding the diagnosis and management options. Early detection also allows time to prepare for a safe delivery in a medical center that offers neurologic closure. Functional motor outcome can also be predicted by high-resolution ultrasound before delivery.39 Previous consideration has been given to performing cesarean section for all mothers with an affected pregnancy to avoid further damage to neural structures. One study reported a less severe lower extremity paralysis in infants born by cesarean section before the onset of labor but showed no difference in those who received cesarean section after the onset of labor.102 Subsequent studies, however, have found no difference in motor outcome with cesarean section.74,97
Intrauterine Surgical Procedures
It is estimated that more than 400 fetuses have received in utero closure of MMC by open maternal–fetal surgery worldwide.173 Studies involving these patients have suggested that the incidence of shunt-dependent hydrocephalus is significantly reduced, that the brain stem and cerebellum are restored to a more normal anatomic configuration, and leg function might be improved in some patients receiving intrauterine MMC repair.45,173,174,186 Criticism exists, however, regarding methods of data collection and study design, among other issues.203 A multicenter, randomized, open-label trial is currently underway to further evaluate this procedure and should be completed in 2010. As well, a percutaneous fetoscopic approach to closure has been reported in a few cases with the purported benefit of reduced maternal risk compared with open maternal–fetal surgery.90
Neonatal Management
Back Defect
After an infant is delivered with an open NTD, a sequence of events is set in place to preserve neurologic function, prevent infection, and stabilize cerebrospinal fluid flow. Early closure (within 72 hours of delivery) reduces the risk of infection in the central nervous system.37 Before closure, the open defect must be protected to prevent contamination or further damage from trauma. Closure occurs in three stages:
After closure of the open spinal defect, hydrocephalus often develops.3 Some centers advocate simultaneous back closure and insertion of a ventriculoperitoneal (VP) shunt (Figure 54-3).103,117
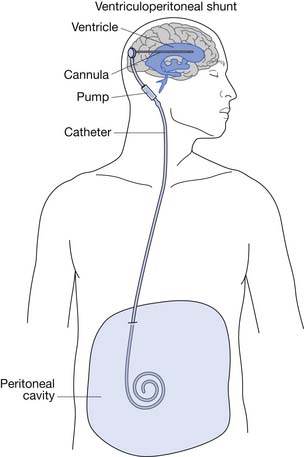
Hydrocephalus
Most infants with MMC require VP shunting.106 Approximately 15% are born with severe hydrocephalus and require immediate shunting.111 The 85% who do not require immediate shunting should be watched closely after their back closure for signs of increased intracranial pressure. The leaking cerebrospinal fluid serves as a decompression before closure, and once the defect is closed, cerebrospinal fluid can accumulate in the ventricular system. The white matter of a neonate is relatively compliant, and therefore the ventricles can become enlarged before the head circumference changes.13 The presence of hydrocephalus correlates well with the level of the spinal defect, with thoracic lesions having a higher incidence than lumbar or sacral lesions.143
Early Bladder Management
More than 90% of infants with MMC will have a neurogenic bladder. Management decisions made in infancy can affect renal health and the eventual development of urinary continence.166 The importance of aggressive urinary management should be stressed to the family before the child leaves the hospital. Early goals include avoiding infections, preventing upper tract damage, and identifying anatomic abnormalities in the genitourinary system.166 Baseline investigations should include renal–bladder sonography and a voiding cystourethrogram. Hydronephrosis is found in 7% to 30% of infants, and reflux occurs in approximately 20% of infants.77,205 Infants with hydronephrosis or reflux should be started on prophylactic antibiotics. Infants who are unable to void begin intermittent catheterization programs. If the infant is able to void, he or she should be checked for complete emptying by checking a postvoid residual volume, either by catheterization or bladder scan. Incomplete emptying can lead to urinary tract infections because the retained urine serves as a culture medium.
Assessment of the Neurologic Level
Even if not spoken out loud, the first question parents of a newborn with MMC often ask is, “Will my child be able to walk?” A careful neurologic examination can give them an idea even within the first few days of life. The best predictor of motor function is the actual motor examination. Information regarding the best motor examination can be obtained by observation, palpation, and postural changes. Motor examinations can improve after the initial examination, which might be related to a period of spinal shock associated with the delivery or the closure.56,160 This can make a newborn’s motor function appear worse than it might eventually be.
Therapy
The goal of any MMC team should be to develop and implement a comprehensive plan that enables the child to attain a maximal level of function in all areas.119 Therapists play a key role in this endeavor and often develop a very good working relationship with families of children with MMC. Therapists are invaluable in providing education and anticipatory guidance for the family. For children born with contractures at the hips, knees, ankles, or feet, a program of passive range of motion can be taught to the family even before patient discharge. Splints are often fabricated soon after birth by the therapists. Throughout life, the MMC clinic therapist will help to educate and coordinate with community therapists regarding the plan developed by the MMC team.
Childhood Management
Shunts
Almost all children with MMC require placement of a VP shunt for management of hydrocephalus. The two most common shunt complications are infection and obstruction.185 Presenting signs and symptoms of shunt malfunction vary with the age of the child. Mechanical obstructions tend to present more acutely with signs and symptoms related to increased intracranial pressure, and infections tend to present more insidiously (Box 54-2).
Infections have a greater long-term morbidity than malfunctions. The overall risk of shunt infection is 12% per child. Staphylococcus epidermidis is the organism that causes most shunt infections.55 Symptoms do not usually develop until several weeks after the shunt is placed or revised. Epidemiologic factors seem to influence the incidence of shunt infections more than surgical factors. Aside from skin contamination during shunt placement, shunts can also become infected with gram-negative rods if the distal end of the shunt erodes into an intraabdominal organ. Gram-negative infections have a much poorer prognosis.57
In the first year of life half of all children with a VP shunt develop obstruction requiring revision. Of those children who require a revision in the first year, 31% will require a second revision in the second year, and then have a risk recurrence rate of 12% per year thereafter.170 Endoscopic third ventriculostomies are performed in carefully selected patients and can become an alternative to chronic VP shunts.23
Arnold–Chiari II Malformations
The Chiari II malformation is characterized by variable displacement of cerebellar tissue into the spinal canal, accompanied by caudal dislocation of the lower brain stem and fourth ventricle (Figure 54-4). Although these posterior fossa abnormalities are the most often described, the Chiari II malformation is also associated with a wide range of abnormalities throughout the neuraxis.3
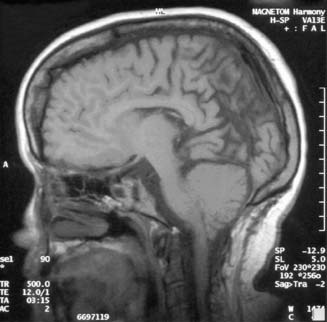
Although the operative mortality for closure of the spinal defect in children with MMC is very low, the operative mortality for symptomatic Arnold–Chiari malformations is relatively high (34% to 38%).3,150 A symptomatic Chiari II malformation remains the leading cause of death for infants with MMC.171 Signs and symptoms of symptomatic Chiari II malformations include intermittent obstructive or central apnea, cyanosis, bradycardia, dysphagia, nystagmus, stridor, vocal cord paralysis, torticollis, opisthotonos, hypotonia, upper extremity weakness, and spasticity. The constellation of stridor, central apnea, and aspiration is sometimes referred to as central ventilatory dysfunction.71
Before hindbrain decompression for a symptomatic Chiari II malformation, the child’s shunt system should be evaluated carefully because shunt malfunctions can cause Chiari malformations to become symptomatic. Hindbrain decompressions should be performed early to minimize the progression of symptoms of the Chiari malformation. Poor preoperative prognostic signs include bilateral vocal cord paralysis, severe neurogenic dysphagia, and prolonged apnea.36
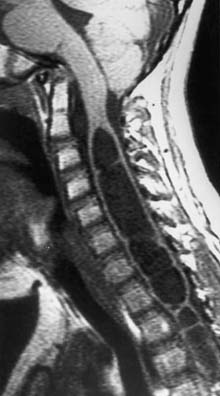
Hydromyelia
Hydromyelia is the dilatation of the central canal of the spinal cord (Figure 54-5). It is analogous to dilatation of the ventricles in the brain and is a relatively common occurrence in children with MMC. Hydromyelia is probably much more common than we are aware because it often does not cause obvious symptoms in patients with MMC.27 When symptomatic, it usually presents with rapidly progressive scoliosis, a change in strength or coordination of the upper or lower extremities, and spasticity. Magnetic resonance imaging is the best study to demonstrate this spinal cord abnormality.26 When suspected, the entire neuraxis should be imaged because untreated or subclinical hydrocephalus can produce hydromyelia. Hydrocephalus should be treated before surgical treatment for hydromyelia.
Tethered Cord Syndrome
In children with MMC, the spinal cord can be fixed or “tethered” at one point, causing traction, which can lead to progressive urologic, orthopedic, or neurologic decline. It was first described in 1857, and the first known detethering of the spinal cord was performed on a previously healthy 17-year-old subject who had progressive loss of lower extremity function in 1891.111
The spinal cord usually terminates at the level of L1–L2. However, MMC repair invariably is followed by the development of arachnoiditis, fibrosis, and adhesions between the intraspinal neural structures, the meninges, and the surrounding vertebral structures.206 These adhesions can tether the cord to the low lumbar or sacral region.
Most children with MMC will show signs of tethering on magnetic resonance imaging. Therefore symptoms should develop before surgical correction is pursued. Typical signs and symptoms in children include increased weakness (54%), worsening gait (54%), scoliosis (51%), pain (32%), orthopedic deformity (11%), and urologic dysfunction (6%).78 Surgical correction should be considered early because most cases will improve or stabilize if treated early.78 Delayed correction can result in irreversible loss of function because the natural history is for symptoms to worsen with time.138
At least three other lesions can lead to tethering of the spinal cord: diastematomyelia, lipomyelomeningocele, and tight filum terminale. Diastematomyelia refers to divisions (not duplications) of the spinal cord. It is usually associated with a bony spur. Even if asymptomatic, the natural history is for symptoms to develop that can be irreversible.5,69 Lipomyelomeningocele refers to a subcutaneous lipoma, continuous with the cauda equina, which also has a meningocele with neural elements enclosed extending outside the dura. A tight filum terminale is another congenital malformation in which the filum terminale does not elongate. Prophylactic surgery is usually recommended for these three lesions.
Neurogenic Bladder
Urologic involvement in MMC and other spinal dysraphisms varies, and is not necessarily correlated with the level of the lesion as in traumatic spinal cord injury. In MMC, more than 90% of patients have partial or complete denervation of the bladder, with poor compliance and contractibility resulting in unacceptable residual urine volumes.166 The urethral sphincter is incompetent in 86% of patients, so that incontinence occurs with increases in intravesical pressure. About one third of patients have detrusor–sphincter dyssynergia, resulting in high intraluminal pressures.109 The external sphincter is usually partially functional and can improve in the first year after birth.168 Patients should be observed at least annually because deterioration or improvement can occur in the first year of life, and tethering of the spinal cord with a change in bladder function can occur over the years. Although the majority of individuals with MMC have normal renal function at birth, 40% to 90% will experience a decline by age 10 if left unattended.164
Prevention of damage to the urinary tract and continence are the primary goals of neurogenic bladder management. Urodynamic evaluation is generally performed in infants with MMC, with 75% showing a normal upper urinary tract. The remaining infants show some degree of hydronephrosis resulting from vesicoureteral reflux, detrusor–sphincter dyssynergia, an enlarged bladder, or other structural abnormality. Infants with normal anatomy should receive a renal ultrasound biannually. Those with incomplete emptying and no outlet resistance can be taught the Credé maneuver. Those with detrusor–sphincter dyssynergia, or who have already developed hydronephrosis, should be treated with anticholinergic medications and clean intermittent catheterization to prevent the development or worsening of hydronephrosis. Children with vesicoureteral reflux, which develops when detrusor pressure exceeds 40 mm Hg, are often prescribed prophylactic antibiotics. If they have persistent febrile urinary tract infection or persistent hydronephrosis, surgical intervention is often necessary. Cutaneous vesicostomy can be performed, with reversal done at a later time when the patient is capable of effective clean intermittent catheterization.110,158
As fewer than 10% of children with MMC have normal urinary control, continence of urine is a prominent issue.96 Although there are no effective external collection devices for girls, excluding diapers, condom catheters are an option for boys with reflex emptying who do not have vesicoureteral reflux or large residual volumes. Appropriate sizing can be a difficult issue for some, and impaired sensation can lead to skin breakdown.
The high prevalence of small bladder capacity and low outlet resistance in many children results in only about one fourth of children being continent with clean intermittent catheterization alone.187 The addition of anticholinergic medications, α-adrenergic agonists, and antibiotic instillations still only resulted in complete continence in 49% of patients in one study.198 In general, frequent catheterization less than every 4 hours is required to achieve continence.96
Multiple surgical options are available for those who do not achieve continence with clean intermittent catheterization and medications. Although a full description of these techniques is beyond the scope of this chapter, bladder augmentation along with artificial sphincter placement can be used individually or in combination. Success for long-term continence after artificial sphincter placement is more than 60%.17 In addition, for patients who have difficulty performing urethral clean intermittent catheterization, continent diversion, with the appendix used as a conduit to the bladder to create an abdominal stoma, can create easier access for many patients.158
Independence with toileting in children with MMC is delayed more than all other self-care tasks, regardless of intelligence. Although most children achieve independent control of bowel and bladder function by the age of 4, those with MMC might not achieve this until age 10 to 15.134 The cause of this is multifactorial and includes level of paralysis, intelligence, difficulty with visuospatial tasks, kyphoscoliosis, parental support, sensation, sphincter control, and bladder perception. Children can be taught to perform clean intermittent catheterization as early as age 5, although they will still need assistance with maintaining a schedule. Parents need to be trained not only in their child’s bladder program but also instructed in the importance of allowing the child to accept responsibility once she or he is able. A recent study of young adults revealed that up to 60% reported urinary incontinence, with approximately 70% perceiving this as a problem.167,195
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


