CHAPTER 87 Metabolic Bone Disorders of the Spine
Cellular Biology
Bone Cells
Four families of cells combine to create bone tissue that is biomechanically functional and structurally sound. These include the osteoprogenitor cell; its derivative, the osteoblast; the incorporated mature form, the osteocyte; and, finally, the osteoclast. Together, these cells produce new osseous tissue. This tissue is maintained through resorption and remodeling. In short, the cells determine mechanical stability and mineral homeostasis. Healthy bone metabolism results from equilibrating these competing forces. Some forms of metabolic bone disease, therefore, can be viewed as the pathologic imbalance—in number, power, or degree of differentiation—of bone-forming and bone-resorbing cells.1,2
The osteoprogenitor cells are components of the bone marrow stromal system, approximating the surfaces of bone (periosteum and endosteum) and the adjacent bone marrow environment.3 These potentially mitotic cells, under appropriate stimulation and conditions, give rise to either the bone-forming osteoblast or the cartilage-producing chondroblast. The natural history of these osteoprogenitor cells and the precise signals for their differentiation and modulation are not yet fully elucidated.
Osteoblasts sit on the metabolically active surface of bone in an adherent row, where they synthesize and release unmineralized bone matrix called osteoid.4–7 They subsequently participate in the mineralization of the osteoid by releasing packets of ions, as well as by synthesizing regulatory and crystal-nucleating noncollagenous proteins. These cells are characterized by high levels of alkaline phosphatase, the ability to manufacture type I collagen and osteocalcin, and the presence of numerous receptor sites for parathyroid hormone (PTH), estrogen, and other products. Osteoblasts are also thought to govern the actions of osteoclasts (described later), thus regulating and coupling bone formation and bone resorption.
The osteocyte, the terminal cell of the osteogenic cell line, is derived from osteoblasts.4,5 As the osteoblast synthesizes bone matrix, it becomes embedded within its product. The cell is now termed an osteocyte, and the space in which it sits is termed a lacuna. There is one osteocyte per lacuna. Despite being surrounded by bone matrix, the osteocyte communicates with other cells via slender cell processes that it extends for some distance into canaliculi in the surrounding matrix. Thus osteocytes are not completely isolated in their lacunae from other cells but appear to communicate with one another and with surface cells via these cell-to-cell junctions. In this way, osteocytes maintain the matrix that envelops them, regulate local ionic concentrations, and govern the degree of mineralization. In addition, they monitor the local mechanical load and signal changes in the matrix. The surface area covered by the canaliculi is greater than the surface area of osteoblasts and osteocytes combined.
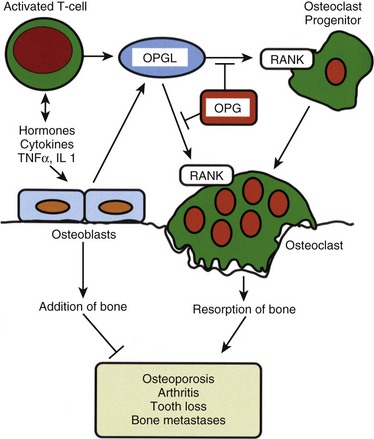
Osteoclasts resorb bone.8,9 Unlike the mesenchymally derived osteoblast lineage, osteoclasts arise from the monocyte line. These large multinucleated cells are found on the resorbing surfaces of bone, where they bind to a bone-specific integrin10 and form an isolated macrocavity. At this attachment cavity site, an acidic microenvironment causes the dissolution of the hydroxyapatite mineral and releases acidic proteases that degrade the organic collagen matrix. The resultant resorption cavity is called a Howship lacuna. Thus by remodeling formed bone and delivering calcium into the circulation, the osteoclasts participate in both the mechanical and biochemical roles of bone tissue. Osteoclasts are devoid of PTH receptors, even though they functionally respond to this hormone. Indeed, the hormonal response is found only in those osteoclasts approximated to functional osteoblasts (which do possess PTH receptors). On stimulation by PTH, osteoblasts produce RANKL (receptor activator of NFκB ligand), which interacts with RANK receptors on osteoclast progenitor cells, leading to the activation of mature osteoclasts. Osteoprotegerin (OPG, also known as NFκB) blocks this pathway. RANKL, RANK, and the decoy receptor OPG are the key regulators for the development and activation of mature osteoclasts (Fig. 87–1). Agents that inhibit RANKL via OPG or anti-RANKL antibodies can be used for the treatment of osteopenic disorders such as osteoporosis as well as bone loss associated with bone metastases.11–14
Bone Matrix
In addition to the cellular component, two matrices—an organic matrix and an inorganic matrix—constitute the remainder of bone tissue (Fig. 87–2). Whereas the inorganic matrix consists of bone mineral, the organic matrix consists of collagenous fibers embedded in a ground substance.
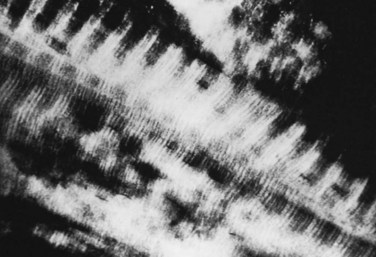
FIGURE 87–2 Collagen-mineral relationship. The hydroxyapatite forms in the hole zone between collagen molecules.
(From Anatomy II. Orthop. Science. Park Ridge, IL, AAOS, 1986.)
Bone Mineral
The inorganic matrix represents two thirds of the dry weight of bone and primarily consists of calcium phosphate,15 which exists in three forms: (1) crystalline hydroxyapatite [Ca10(PO4)6(OH)2], the most abundant; (2) octacalcium phosphate [Ca8H2(PO4)6 · 5H2O)], a rarer form; and (3) brushite [CaHPO4 · 2H2O]. The hydroxyapatite forms platelike crystals 40 nm in length and 3 nm in thickness. Occasional contaminants including carbonate substituted for a phosphate or a fluoride for a hydroxy group (as well as the potential inclusion of lead or arsenic) disrupt the purity of the crystals. These contaminants alter the physical properties of the matrix and may also affect the biologic characteristics. The mineral crystal closely associates with the organic matrix, initially deposited in the hole zones of the collagen fibril. Later, it surrounds itself with ground substance (proteoglycan), as well as with water and other ions.
Collagen
Concentrations of calcium and phosphate alone do not dictate mineral deposition. An organic matrix, consisting of collagenous fibers embedded in ground substance, assists with and regulates mineralization.16 The organic matrix of bone is primarily composed of proteins, approximately 95% of which is collagen in adult humans. Collagen, a rigid macromolecule, employs tropocollagen as its basic structural unit. Tropocollagen, in turn, is composed of three polypeptide chains, each comprising approximately 1000 amino acids.17 In bone collagen (type I), two of these chains share an identical amino acid sequence; a third chain (α2) has a similar, but not identical, sequence. All three chains contain unusually high concentrations of glycine, proline, alanine, hydroxyproline, and hydroxylysine and are wound into right-handed helices.
Type I collagen synthesis occurs in osteoblasts. It is formed as a precursor “procollagen” molecule in the cytoplasm and then extruded from the osteoblast after cleavage of signal sequences. Numerous additional modifications are subsequently made to the secreted procollagen molecule. Notably, extensive nonhelical regions are removed from both its amino and carboxy terminals, and key proline and lysine residues are hydroxylated by an iron-containing enzyme. (The iron in this hydroxylase functions only in the ferrous state; therefore ascorbic acid, functioning as a reducing agent, is necessary.) The hydroxylation of proline and lysine occurs after these amino acids are incorporated into the chain; therefore free hydroxyproline in the serum signifies lysis of collagen or its precursors. Accordingly, measurements of urinary hydroxyapatite excretion provide a qualitative measure of bone turnover. The hydroxylation of proline and lysine assists crosslinking, which, in turn, lowers the solubility of collagen and increases its tensile strength. Collagen is one of the strongest components of bone; the tensile strength of bone collagen exceeds the compressive strength of the bone mineral by 2000 N/cm2. Pyridinoline, deoxypyridinoline, C-telopeptides, and N-telopeptides are crosslink breakdown products that all provide better markers of bone resorption than hydroxyproline.18,19 They are elevated in high-turnover osteoporosis and Paget disease, and they decline rapidly with successful antiresorptive therapy (see later). Adequate nutrition and vitamin C (ascorbic acid) are required for collagen maintenance.
Type I collagen is found not only in the bones of the spine but also in the intervertebral discs, where it coexists with type II collagen. These homologous molecules differ only in the chains that compose their triple helices. Within the disc, each type of collagen maintains a separate anatomic domain: type I collagen is more abundant in the annulus fibrosus, whereas type II predominates in the nucleus pulposus.20
Ground Substance
Proteoglycans form the ground substance of bone. A proteoglycan monomer is composed of two kinds of glycosaminoglycans—keratin sulfate and chondroitin sulfate—joined to a core protein. Keratin and chondroitin sulfate form through the polymerization of disaccharide units, one sugar of which is always a hexosamine possessing an anionic group. This negatively charged group binds water, expanding the tissue volume and imparting resilience to the ground substance. In addition, the presence of water in the ground substance allows for the diffusion of metabolites in the organic matrix. Thus the proteoglycans in bone exert regulatory and structural forces. When the bone mineralizes, the water content is reduced, leading to denser, more compact tissue. The intervertebral discs also contain proteoglycans, which comprise 30% to 60% of the dry weight of the nucleus pulposus. With age, proteoglycan concentration (and, consequently, water concentration) declines. However, even with decreased proteoglycan concentration, osmotic forces allow the proteoglycans to remain somewhat hydrated.21
Osteocalcin is a bone-specific protein that, like type I collagen, is synthesized by osteoblasts.22 Osteocalcin comprises only 2% of the organic matrix by weight. Vitamin D enhances the synthesis of osteocalcin but is not an absolute requirement for synthesis. Osteocalcin prefers to bind to calcium within hydroxyapatite. The precise function of this prevalent osseous protein is unknown, although its levels are correlated with bone mineral content. Possible roles include the attraction of osteoclasts to sites of bone resorption, regulation of the rate of mineral turnover, and determination of the morphology of the mineral crystal. Osteocalcin, derived from new bone synthesis, circulates in plasma at concentrations proportional to osteoblast activity. A correlation between abnormal osteocalcin levels and radiographic evidence of metabolic bone disease has been suggested.
Osteonectin, a 32-kd phosphoprotein secreted by osteoblasts, is the second most prevalent protein in bone. It binds to both collagen and hydroxyapatite,23 as well as to carbohydrate moieties. Putative roles for osteonectin include calcium phosphate nucleation and stabilization of the newly formed crystal.
Bone morphogenetic protein (BMP), as the name implies, induces osteoprogenitor cells to form bone.24 A small protein, BMP accounts for only 0.1% of the total bone protein mass in cortical bone, and even less in the spine. More than 15 BMPs exist, and some are related to the transforming growth factors. Experimentally, BMP implanted in ectopic sites (in the absence of bone collagen) induces perivascular mesenchymal cells to become bone-forming osteoprogenitor cells. The absence of BMPs (or the inhibition of their activity) may contribute to the pathogenesis of diseases of decreased bone mass or impaired bone remodeling or repair. The genes for the expression of BMPs have been cloned,24,25 and the protein has proven to possess a pharmacologic role in repairing bone and inducing spinal arthrodesis.26
Skeletal Homeostasis
Bone growth occurs during all phases of life.4,5 Functionally, this growth can be divided into two processes: modeling and remodeling. The deposition of new bone in regions not first cleared by resorption or the resorption of old bone without deposition of new tissue is termed modeling. Modeling results in changes to the external shape, mass, or volume of existing bone. Alternatively, remodeling retains approximately the external form of bones. During remodeling, bone replaces itself by balancing synthesis in some places with lysis in others. This balanced turnover releases ions into the circulation and prevents accumulation of aged or fatigued bone.
Mineral Homeostasis
Calcium
Bone exchanges calcium, magnesium, and phosphate and participates in acid-base balance. Of all of these, calcium homeostasis is the most important to both the skeletal and metabolic functions of bone.27–30 Calcium fulfills its skeletal role in the hydroxyapatite mineral crystal, where it provides mechanical strength. It serves its metabolic function as a free divalent cation. These functions include transducing hormonal signals within the cytoplasm (“second messenger” function), coupling neural excitation with muscular contraction, and effecting homeostasis by interacting with both vascular smooth muscle and platelets.
Active absorption in the duodenum takes place by means of a protein-dependent transport system31 that is activated by 1,25-dihydroxyvitamin D (1,25[OH]2D). In the jejunum, passive diffusion accounts for the remainder of the calcium absorbed. Clearly, the jejunal phase depends more on concentration gradients and intestinal transit times. In addition, dietary composition affects absorption: Diets high in fiber and oxalate (both found in green vegetables) allow for less calcium absorption. In times of increased calcium demand such as the growth years, or during pregnancy, the duodenal fractional absorption can be augmented by increased carrier protein synthesis, which is in turn stimulated by the active vitamin D metabolite (see later). The efficiency of calcium absorption decreases with aging and is adversely affected with bariatric surgery.29,32
Phosphate
The second important ion in bone metabolism is phosphate.33 Like calcium, phosphate serves multiple functions throughout the body. As a component of adenosine triphosphate, phosphate participates in the interconversion of the energy of metabolism; as a constituent of nucleotides, phosphate partakes in the transmission and expression of genetic information; and in 2,3-diphosphoglycerate, it regulates the oxygen affinity of hemoglobin.
As with calcium, the kidney is responsible for maintaining phosphate balance,34 and it does so by a similar mechanism: proximal sodium bulk-driven flow and distal control. When phosphate intake is high, the kidney excretes increasing amounts by spilling the excess phosphate into the urine. When serum phosphate concentrations are low, the kidney can avidly conserve phosphate by implementing vitamin D–mediated changes in calcium concentration. In addition to calcium balance, dietary load, volume status, and acid-base balance also affect renal phosphate handling.
Regulators of Bone and Mineral Metabolism
Parathyroid Hormone
Parathyroid hormone is an 84-amino acid polypeptide secreted by the chief cells of the parathyroid glands in response to low serum calcium.35 Its physiologic role is to restore a normal calcium concentration by stimulating all three organs of calcium homeostasis. The kidney and the bone are affected directly,29,36 whereas the intestine is affected only indirectly, by means of the synthesis of the active vitamin D metabolite 1,25(OH)2D3.
PTH promotes calcium conservation in the kidney.37 There, increased serum calcium is achieved through a twofold mechanism; both steps depend on stimulation of adenylate cyclase, the enzyme that forms cyclic adenosine monophosphate (cAMP). First, PTH increases calcium resorption in the distal nephron. In addition, it promotes the loss of phosphate. This phosphaturic effect prevents the recently resorbed calcium from being deposited into the bone hydroxyapatite or into ectopic calcium-phosphate deposits. Moreover, the excretion of phosphate lowers the levels of circulating calcium-anion complexes, causing more of the serum calcium to remain in the physiologically useful, free form.
In the bone, PTH affects calcium metabolism by increasing the surface resorption of the mineral by osteoclasts, by promoting ion flux by osteocytes, and by decreasing their bone synthetic activity through the inhibition of calcium “consumption” by osteoblasts. PTH increases the number of resorptive surfaces in bone and, within them, the density of osteoclasts. The precise mechanism of this osteoclastic action is unfolding and appears dependent on the actions of osteoblasts. The osteoblast does possess PTH receptors. It is postulated that PTH induces the osteoblast to activate the RANKL-RANK system, which stimulates osteoclastic bone resorption. The catabolic effects are reversed by low, intermittent doses of PTH that directly stimulate osteoblastic bone formation but do not activate RANKL.21
Vitamin D
Unlike the polypeptide hormone PTH, vitamin D is a steroid.36 As such, it defies rapid proteolytic inactivation and thus functions best as a longer-acting regulator of calcium homeostasis. The principal effect of this hormone is to increase intestinal absorption of calcium from the diet, a process that is usually only 25% efficient. Secondarily, it complements PTH in the promotion of calcium resorption from the bone, again by promoting transport across cell membranes (in this case, bone cells rather than intestinal cells).
Cholecalciferol (inactive vitamin D3) forms in the skin from the ultraviolet light activation of 7-dehydrocholesterol, an intermediate of cholesterol synthesis.38 7-Dehydrocholesterol can degrade and re-form cholecalciferol; therefore once formed, cholecalciferol is removed from the local environment by a transport protein, thus favoring further formation. As little as 15 minutes of sunlight exposure allows the synthesis of a fair-skinned individual’s vitamin D3 requirement. The total need for sunlight increases with melanin concentration. In the absence of sun exposure, cod liver oil and enriched milk and cereals can provide usable precursors of the hormone.
In the liver, vitamin D3 undergoes 25-hydroxylation,39 forming the prehormone 25-hydroxyvitamin D (25[OH]D). Subsequent 1-hydroxylation to the active form, 1,25(OH)2D3, takes place in the kidney and is promoted by PTH. In the presence of low PTH, an alternative hydroxylation occurs at the 24 position, yielding 24,25(OH)2D3, an inactive form of the hormone. Unlike PTH, which is functionally regulated at the level of secretion (calcium levels do not cause minute-to-minute changes in messenger RNA activity but rather control the release of PTH), vitamin D is regulated at the level of biosynthesis. The 1-hydroxylation reaction is the rate-limiting step and is regulated by feedback inhibition, as well as by PTH levels.
Active vitamin D, also known as calcitriol, promotes increased transport across cell borders. As a steroid maturation hormone, vitamin D works by crossing the cell membrane and subsequently entering the nucleus. There it binds to DNA to promote DNA translation, thus increasing the synthesis of the target proteins. Vitamin D mediates increased expression of calcium-transport proteins, although the nucleic receptor has not yet been identified. In the gut, 1,25(OH)2D3 causes increased calcium transport; phosphate absorption increases as well. In the bone, 1,25(OH)2D3 complements the action of PTH by increasing calcium transport across bone cell membranes, thus assisting with the mobilization of calcium from the bone and into the circulation. As such, it functions as an agent of bone resorption. It can indirectly assist with bone formation as well because the tasks of mineral homeostasis and skeletal homeostasis are not always at odds with each other. Calcium needs can usually be met through the diet, not by bone resorption. Vitamin D also functions as a maturation hormone by increasing the villous membrane of the gut and augmenting PTH recruitment of macrophage stem cells, the bone-resorbing osteoclast precursors. Accordingly, vitamin D, which promotes the uptake of dietary calcium, typically also serves as an agent of bone maintenance. In addition, vitamin D has been associated with decreased cancer rates, improved muscle function, and a more normal immune system.40
Calcitonin
Calcitonin is a 32-amino acid polypeptide secreted by the parafollicular cells in the thyroid in response to increased serum calcium concentration. Calcitonin lowers serum calcium levels by its actions in the bone and the kidney. Still, its precise physiologic role awaits determination.41,42
In the bone, calcitonin rapidly inhibits osteoclastic action at pharmacologic doses. Direct calcitonin-binding sites are present on osteoclasts. Calcitonin decreases the number and activity of osteoclasts. Specifically, it decreases the adherence of osteoclasts to bone resorptive surfaces and diminishes their activity once they adhere. Calcitonin also affects osteocytes, causing decreased calcium ion flux across their cell membranes. In the kidney, calcitonin blocks the reuptake from the glomerular fluid of calcium and phosphate, as well as that of other ions, notably sodium (which influences calcium uptake by the passive mechanisms described earlier). Calcitonin may also influence calcium handling in the gut and, at pharmacologic doses, may alter other intestinal processes (such as water balance).43
Osteoporosis
Osteoporosis is a metabolic bone disease characterized by low bone mass and microarchitectural deterioration of bone tissue, leading to increased bone fragility and a consequent increase in fracture risk, especially of the hip, spine, and wrist. Osteoporosis affects an estimated 28 million Americans. Of these, 10 million are estimated to already have the disease and nearly 18 million more are estimated to have low bone mass, placing them at increased risk for osteoporosis.44 It is the most prevalent metabolic bone disease. The estimated national direct expenditures (hospitals and nursing homes) for osteoporotic and associated fractures were $17 billion in 2001 (or $47 million each day) and will likely rise with the expanding elderly population. In postmenopausal women and in the elderly, osteoporosis is a major underlying cause of pathologic fractures. At least 1.5 million fractures each year are attributable to the disease,45 and almost half of these occur in the spine. Moreover, one third of all American women older than 65 years of age suffer vertebral fractures.46
Like all metabolic bone diseases, osteoporosis is not limited to the spine. In addition to the annual 700,000 vertebral fractures caused by osteoporosis, the disease claims 300,000 hip fractures and 250,000 Colles fractures.45 In the elderly, hip fractures, most caused by osteoporosis, result in death, disability, and dependency: The mortality rate of older patients with hip fractures can be as much as 20% higher than in persons of similar age47; up to 50% require long-term nursing home care, and less than 30% return to a lifestyle comparable with their prefracture state.48 Among the vertebral fractures, two thirds are clinically silent; however, these patients have lower quality-of-life function.49 Women with at least one new vertebral fracture had an age-adjusted 32% increased risk of mortality. When these women were stratified by number of incident vertebral fractures, those with two or more incident vertebral fractures had a 1.47-fold increased risk of mortality.50
Generally, osteoporosis is classified as either primary or secondary. Primary osteoporosis is further subdivided on the basis of its pathogenesis. Type I, or postmenopausal osteoporosis, is related to the abrupt decline of estrogen levels that occurs in menopausal women. Type II osteoporosis, known as senile or age-related osteoporosis, is due to the progressive decrease of bone mineral density in both men and women that occurs with aging. Patients may suffer from both subtypes of primary osteoporosis. Primary osteoporosis can occur in both men and women of all ages but often follows menopause in women and occurs later in life in men. Several risk factors for osteoporosis have been identified. A typical patient is a slim, white, postmenopausal woman of northwestern European descent.38 She may have a history of premature menopause, cigarette or excessive alcohol use,27 an eating disorder (e.g., anorexia nervosa), a sedentary lifestyle, use of anticonvulsants,51 or lifelong low calcium intake. Seventy-five percent of women with scoliosis older than 65 years of age have at least one osteopenic wedge fracture.52
Secondary osteoporosis results from any medical condition or medication that contributes to accelerated bone loss. The incidence of secondary osteoporosis is higher in men than in women and is estimated to be 30% to 60%.53 Causes of secondary osteoporosis include endocrine and gastrointestinal disorders, bone marrow dysplasias, and disorders of calcium balance, among others.54,55 Endogenous hyperthyroidism increases cortical bone loss and may accelerate skeletal deterioration.56 Inflammatory bowel disease (i.e., Crohn disease or ulcerative colitis) can cause osteoporosis both from the disease itself and its treatment. Malabsorptive diseases of the gut such as celiac sprue are also associated with low bone density and osteoporosis. Corticosteroid-induced osteoporosis is the most common cause of drug-related osteoporosis and is associated with a high fracture rate. Corticosteroid therapy causes bone loss and fractures because it suppresses bone formation and inhibits intestinal calcium absorption, which leads to secondary hyperparathyroidism and increased osteoclastic bone resorption.57 In addition, osteomalacia (vitamin D deficiency) may mask itself as osteoporosis. The distinction is important because antiresorptive medications for osteoporosis are not appropriate choices to treat osteomalacia. Many secondary causes of osteoporosis are treatable and may necessitate a different management course than primary osteoporosis.
Pathology
Osteoporosis is a disease of decreased bone mass in the absence of a mineralization defect. Bone mass is diminished; however, the bone that remains contains a normal matrix and a normal degree of calcification. Declining bone mass is now thought to be a universal phenomenon of aging.58 Peak bone mass is attained in the mid 30s for both sexes. Gender, nutrition, race, exercise habits, and overall health all influence bone mass. Peak bone mass is higher in men than in women and higher in African Americans than in whites.59 After the fourth decade, both men and women lose bone mass from the skeleton (Fig. 87–3). Two phases of this loss have been identified: slow and accelerated. The slow phase, related to an imbalance between resorption and formation leading to negative calcium balance, is equal in both men and women. It results in an annual basal slow phase rate of bone loss of 0.3% to 0.5%.60 The accelerated phase that occurs with estrogen deficiency—a phenomenon found exclusively in women—is responsible for cortical bone mass loss of 2% to 3% per year. This loss is in addition to the slow phase losses, which continue during the accelerated phase. The accelerated phase begins after surgical or natural menopause and lasts for approximately 5 years.60 Thereafter, bone loss continues at the basal slow phase rate.
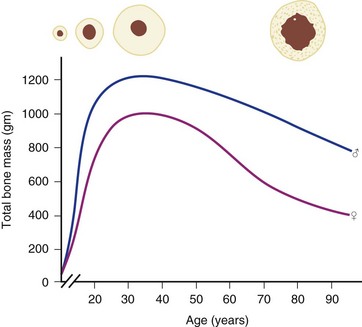
FIGURE 87–3 Bone mass as a function of age in men and women.
(From Disorders of Bone 9. Orthop. Science. Park Ridge, IL, AAOS, 1986.)
Multiple studies have shown the importance of estrogen deficiency in the causation of the accelerated phase.61,62 Estrogen is thought to play a critical role in maintaining bone mass in adult women by suppressing cancellous bone remodeling and maintaining remodeling balance between osteoblastic and osteoclastic activity. With estrogen deficiency, there is increased osteoclastic activity and possibly also impaired osteoblastic activity, resulting in a remodeling imbalance.63 The administration of estrogen to women during this period of rapid bone loss can decrease the loss in all bones, especially those rich in trabecular bone (e.g., the vertebral bodies, the pelvis and other flat bones, in the ends of long bones).64
Riggs and Melton60 have subclassified primary osteoporosis on the basis of patterns of bone loss and fracture (Table 87–1). Type I or “postmenopausal” osteoporosis primarily affects trabecular bone sites and is characterized by fractures of the vertebral bodies and wrist. In patients with type I osteoporosis, the rate of trabecular bone loss is three times above normal but the rate of cortical bone loss is only slightly above normal. In contrast, type II osteoporosis, also known as “senile” osteoporosis, occurs in both men and women aged 75 years and older and involves areas of predominantly cortical bone. Clinically, fractures of the hip, pelvis, proximal humerus, and proximal tibia are seen. The causes of senile osteoporosis are the aging process itself and chronic calcium deficiency. Senile osteoporosis may also involve decreased vitamin D and increased PTH activity or impaired bone formation. Osteoporosis in patients between the ages of 66 and 74 years may represent a combination of these two syndromes.
TABLE 87–1 Type I versus Type II Osteoporosis
| Type I | Type II | |
| Female-male ratio | 6 : 1 | 2 : 1 |
| Calcium deficiency | No | Yes |
| Estrogen deficiency | Yes | No |
| Bone loss | Disproportionate loss of trabecular bone | Proportionate loss of both cortical and trabecular bone |
From Riggs BL, Melton LJ 3rd: Evidence for two distinct syndromes of involutional osteoporosis. Am J Med 75:899, 1983.
The spine is composed of primarily trabecular bone. Compared with cortical bone, it has a high surface-to-volume ratio. Because metabolic activity (remodeling) occurs on bone surfaces, trabecular bones in general, and the vertebral bodies in particular, are resorbed preferentially in times of skeletal loss.65 Osteoporosis is thus characterized by trabeculae of decreased size and number. Work by Dempster and colleagues66 has demonstrated that osteoporosis involves a thinning of the cortex, as well as a change in the shape of the trabecular bone from plates to narrow bars. The trabecular bone contains areas in which osteoclasts create a loss of connectivity, leading to a significant and localized weakening of the bone.66,67 Vertebral body density declines before a similar loss is detected in cortical areas.
The body accommodates bone loss by redistribution. As people grow older, the diameter of the long bones gradually increases in both women and in men. Concurrently, the medullary diameter also increases, leading to a net thinning of the cortical bone. A 10% shift of bone mass outward from the epicenter through an enlargement of the bone diameter will compensate for a 30% decrease in the bone mass against applied bending and torque stresses but not against axial loading. This differential resorption explains the timing and patterns of the fracture syndromes seen in osteoporosis.46 The incidence of vertebral compression fractures rises immediately after menopause, whereas the hip (with its higher proportion of cortical bone) fractures later in life, when cortical bone loss accumulates over the next 1 to 2 decades. The distal forearm, like the spine, has high trabecular content, so the incidence of Colles fractures also increases in menopause.
Deficiencies in dietary calcium cause decreased peak bone mass.68 White women on average have less bone than either white men or African Americans of either sex.69 Thus the risk of clinically significant osteoporosis depends on hereditary factors, gender, race, and nutrition, which all contribute to peak bone mass, and aging, which causes progressive bone loss. Although the formation of good bone in sufficient quantity in young adulthood is clearly important, the aging process remains the most important cause of involutional osteoporosis.
Aging leads to bone loss independent of menopause, but the rapid decline of skeletal mass after estrogen deficiency implies that this hormone prevents the dissolution of the skeleton.70 Nonetheless, its precise mechanism is unclear. Some investigators believe that the action is mediated by estrogen receptors on osteoblasts.71 Others contend that estrogen antagonizes PTH activity72 or stimulates endogenous calcitonin release. Decreased calcitonin levels have been found in oophorectomized and postmenopausal patients, and increased calcitonin levels are noted after estrogen administration. Regardless of the mechanism, estrogen deficiency leads to bone loss and plays a major role in the pathogenesis of type I osteoporosis.
The age-related (“slow phase”) bone loss is not affected by estrogen; rather, it likely represents impaired vitamin D metabolism. As an individual ages, the kidney gradually loses its ability to hydroxylate vitamin D into its active form, 1,25(OH)2D.73 This active vitamin D is necessary to transport calcium in the gut; therefore decreased hydroxylation, often coupled with poor dietary intake, leads to lowered serum calcium levels and, in turn, to elevated secretion of PTH. In addition to secondary hyperparathyroidism, elderly people are also more likely to take medications (e.g., diuretics) that further contribute to calcium losses. Furthermore, there is some evidence that the mechanism in remodeling that couples the actions of osteoclasts and osteoblasts functions suboptimally in old age: after the fourth decade, at a given site of remodeling, less bone is laid down than is resorbed.69 Accordingly, normal bone turnover in the elderly leads to calcium depletion as well.
Clinical Course
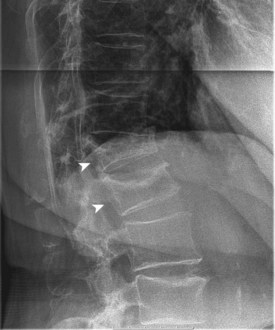
End-stage osteoporosis culminates in fracture.27,29,74 Still, for most of its course osteoporosis is a silently progressive disease. Patients typically present to the physician, usually late in the course, following one of three general scenarios. In the first of these, the patient presents with an acute painful fracture, usually of the spine (Fig. 87–4), but possibly of the rib, wrist, or hip. In the spine, normal activity (even minimal activity) may exceed the depleted bones’ stress tolerance and result in fracture. The acute fracture can be severely painful, with the pain remaining over the affected area or radiating across the thorax. The pain from a vertebral fracture does not radiate down the legs. Symptoms such as leg pain suggest involvement of the spinal cord and obligate the physician to search for another or concomitant process to explain the pain. Osteoporosis, even if established in such a case, is probably not the only disease present. The acute pain usually abates when the fracture heals. Nonetheless, the patient rarely returns to the prefracture status. At a minimum, there may be point tenderness over the fracture site. The patient may note constant abdominal pain, often brought about by the constraining forces on the viscera in the now smaller abdominal cavity. Other patients complain of generalized backache. The backache may be due to changing muscular demands brought on by altered spinal curvature. Furthermore, some patients fear reinjury and strictly limit their activities. Others develop chronic pain syndromes, dysthymic states, or even overt clinical depression. Osteoporosis, therefore, often has a profound effect beyond the acute fracture episode.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


