CHAPTER 37 Medical Myelopathies
Spinal cord disease may result from a variety of insults. A general classification of these various etiologies is provided in Table 37–1. This chapter addresses the “medical” causes of spinal cord dysfunction.
TABLE 37–1 Etiologies of Myelopathy
Metabolic and nutritional disease of the spinal cord: Vitamin B12 deficiency, chronic liver disease, hyperparathyroidism, hyperthyroidism |
Generally, but not invariably, a thorough neurologic examination allows the physician to distinguish spinal cord from other causes of these neurologic complaints. The presence of concomitant neurologic illness attributable to disease of the cerebral hemispheres or brainstem, such as hemianopsia, ophthalmoplegia, and dysarthria, does not rule out the possibility of concomitant spinal cord disease, but should suggest that the findings on neurologic examination may be ascribed to a single or multiple lesions higher in the neuraxis than the spinal cord. The physical examination of patients with spinal cord lesions that have developed gradually or who have acute lesions that have been present for several weeks typically reveals a spastic weakness of the involved extremities with associated hyperreflexia and pathologic reflexes. The latter include Hoffman sign (palmar flexion of the thumb when the distal phalanx of the middle finger of the same hand is rapidly tapped) in the upper extremities and Babinski sign (plantar extension of the hallux when the sole of the foot is stroked) in the lower extremities. Several variants of these signs have been described.1 Superficial reflexes, such as abdominal and cremasteric reflexes, are absent.
Acute Idiopathic Transverse Myelitis and Multiple Sclerosis
Patients with acute idiopathic transverse myelitis most often present with paresthesias of the feet, toes, or fingertips.2 Gradually developing numbness and coincident weakness of the legs follows with subsequent paralysis of the legs. The features of this illness usually evolve over 1 to 3 weeks, but they may develop abruptly. The initial symptoms may be predominantly unilateral, and asymmetrical findings are common. As the illness progresses, upper extremity numbness and weakness and bowel and bladder incontinence may ensue. In some patients, posterior column involvement (decreased vibration and position senses) is spared early but affected later. Occasionally, back (interscapular) pain and, more rarely, calf, arm, or radicular pain may accompany the progressive myelopathy suggesting other pathologic processes, such as an intraspinal neoplasm or epidural abscess. To be considered idiopathic, acute transverse myelitis should be unassociated with a known preceding or concomitant viral infection. The dominant pathologic feature of the spinal cord is demyelination.
Treatment with adrenocorticotropic hormone (ACTH) or corticosteroids has been advocated; the response to therapy may be highly variable, and some studies indicate no benefit.3 A pilot study of high-dose methylprednisolone in children with acute transverse myelitis showed a significant shortening of motor recovery compared with historical controls.4 Approximately one third of patients have a return of a normal gait and bladder function.2 When recovery to a normal or near-normal level of function occurs, it does so within 1 year of the onset of the illness. Of affected patients, 25% become wheelchair-bound or bedridden, and the remainder have varying degrees of lesser disability.2
In some instances, myelopathy develops abruptly. Frequently, a flaccid, areflexic paralysis of the affected limbs accompanies spinal shock. Loss of vision resulting from optic nerve inflammation may accompany this myelopathy. Devic disease (neuromyelitis optica [NMO]) refers to the combination of acute optic neuritis and transverse myelitis. Similar to the more slowly evolving idiopathic transverse myelitis, demyelination is a characteristic neuropathologic hallmark. The pathology has been described as a necrotizing hemorrhagic leukomyelitis. The prognosis of patients with this rapidly evolving myelopathy, particularly when it is accompanied by spinal shock, is worse than the prognosis for the more gradually developing transverse myelitis.5
The differential diagnosis of idiopathic transverse myelitis includes multiple sclerosis; infectious myelitis; postinfectious or postvaccination myelitis; and myelopathy associated with vasculitis and connective tissue diseases, such as Behçet disease, systemic lupus erythematosus (SLE), and Sjögren syndrome. Multiple sclerosis may initially manifest as a transverse myelitis; the likelihood of transverse myelitis being the presenting manifestation of multiple sclerosis was previously reported to be 5% to 15%.2,5–7 Since the advent of more sensitive testing (magnetic resonance imaging [MRI]), this likelihood has been estimated at 42%.8 If cranial MRI is highly suggestive of multiple sclerosis at the time of onset of myelitis, the risk of progression to clinically definite multiple sclerosis is 50% at 2 years, whereas if cranial MRI is negative, the risk is 5%.9 A temporal association with a viral illness (Table 37–2), signs and symptoms that develop over a few days, and a monophasic course should suggest the possibility of a viral myelitis.10
TABLE 37–2 Viral Etiologies of Myelitis
| RNA Viruses | DNA Viruses |
|---|---|
| Nonenveloped | |
| Picornaviruses | Hepatitis B |
| Coxsackieviruses | |
| Echoviruses | |
| Polioviruses | |
| Other enteroviruses | |
| Hepatitis A | |
| Encephalomyocarditis virus | |
| Enveloped | |
| Togaviruses | Herpesviruses |
| Arbovirus | Herpes simplex |
| Rubella | Varicella-zoster |
| Tick-borne encephalitis virus | Epstein-Barr virus |
| Retroviruses | Cytomegalovirus |
| Human immunodeficiency virus type 1 | Herpes simiae |
| Human T-cell lymphotropic virus type I | Poxviruses |
| Orthomyxoviruses | Vaccinia |
| Influenza | Variola |
| Paramyxoviruses | |
| Measles | |
| Mumps | |
| Bunyaviruses | |
| California encephalitis virus | |
| Arenavirus | |
| Lymphocytic choriomeningitis | |
| Rhabdovirus | |
| Rabies | |
Adapted from Tyler KL, Gross RA, Cascino GD: Unusual viral causes of transverse myelitis: Hepatitis A virus and cytomegalovirus. Neurology 36:855-858, 1986.
Spinal cord disease is extremely common in the setting of multiple sclerosis and frequently is responsible for the associated extremity weakness, spasticity, gait abnormalities, and sphincter disturbances. Within 10 to 15 years of the onset of multiple sclerosis, more than 80% of patients exhibit extremity spasticity and weakness, and more than 50% have sphincter disturbances.11,12 Lhermitte sign has been reported in 38% of patients with clinically definite multiple sclerosis.13 Approximately 10% of patients with multiple sclerosis have a primary progressive form of the disease. This disorder often affects the spinal cord predominantly and most commonly develops in women older than 40 years.
NMO has been regarded as a variant of multiple sclerosis; however, the occurrence of a unique IgG antibody to the aquaporin-4 channel (NMO-IgG)14 suggests it is a distinct disorder. NMO may occur as an isolated condition or, more commonly, as recurrent spells of optic neuritis and myelitis. In contrast to the myelitis of multiple sclerosis, which seldom extends beyond two or three segments, the myelitis of NMO is often longitudinally extensive. Optic neuritis or myelitis in isolation may occur with NMO. Treatment recommendations15 include high-dose intravenous methylprednisolone; plasma exchange; intravenous immunoglobulin; and rituximab, an anti-CD20 antibody directed against B cells.
MRI of the involved area of the spinal cord is the best radiographic study. MRI may be normal or reveal cord swelling or hyperintense lesions or both on T2-weighted imaging intrinsic to the spinal cord. Miller and colleagues16 found MRI lesions at the clinically expected level in 64% of patients with a clinical syndrome suggestive of cervical involvement but in only 28% with a suspected thoracic or lumbar lesion. In patients with multiple sclerosis, most lesions are observed in the cervical spinal cord at autopsy and by MRI.17,18 Lesions on MRI are characteristically T2-weighted hyperintense signal abnormalities, generally less than 10 to 15 mm in length,18 and multiple, gapped lesions may be observed. Gadolinium enhancement with active disease is often observed.19 Generally, MRI, if available, negates the need of myelography. If MRI is unavailable, myelography should be performed with water-soluble contrast medium and computed tomography (CT) scan of the affected area. If pain is a significant component of the patient’s illness, a CT scan of the appropriate area and a bone scan may be desirable in eliminating the possibility of an epidural abscess or neoplasm.
Somatosensory evoked potentials can be very helpful in ruling in suspected cord lesions. The presence of abnormalities on brainstem auditory evoked potential and visual evoked potential testing would raise the suspicion of multifocal disease (i.e. multiple sclerosis). Absent F waves on nerve conduction testing can also support evidence of a cord lesion.20
Adrenomyeloneuropathy
Adrenomyeloneuropathy is a phenotypic variant of the genetic disorder adrenoleukodystrophy, in which myelin sheath abnormalities of the white matter are associated with adrenal insufficiency. Although adrenoleukodystrophy is a sex-linked disorder, it has variable expressions, including mild disease in heterozygous women. Pathologically, the white matter abnormality appears to be a diffuse myelinoclastic sclerosis, but it may occasionally appear as a dysmyelinating condition. The neurologic variants are explained by the degree of involvement of brain, spinal cord, and peripheral nerve. Biorefringent material in the adrenal glands and brain observed histopathologically have been shown by sequential extraction methods to be cholesterol esters with high quantities of very long chain fatty acids. Measuring fatty acids in cultured skin fibroblasts from affected individuals, Moser and colleagues21 showed abnormally large amounts of very long chain fatty acids (C24-C30) and a high ratio of C26 to C22 fatty acids. The latter has become the preferred method of diagnosing the disorder.
In patients with the spinal-neuropathic form of this disorder, adrenal insufficiency is usually present since early childhood, and a progressive spastic paraparesis and relatively mild peripheral neuropathy develop in the 3rd decade.22 Other variants affecting the spinal cord have been observed, including a progressive myelopathy in men, a mild spastic paraparesis in women, and combined cerebral and spinal involvement in children and young men. A positive family history is present in approximately 50% of affected individuals. The presence of Addison disease also provides a strong clue to the diagnosis. Addison disease, which is caused by primary adrenal failure, is often accompanied by bronzing of the skin because of excessive secretion of melanocyte-stimulating hormone in association with ACTH.
Infectious Myelopathies
Viral Myelitis
Human Immunodeficiency Virus Infection
Neurologic complications develop in 40% to 60%23–25 of all patients with acquired immunodeficiency syndrome (AIDS), and neurologic disease heralds human immunodeficiency virus (HIV) infection in 10%26 to 20%25 of HIV-infected patients. In retrospective clinical series, spinal cord disease occurring in association with AIDS has been infrequently observed. Levy and colleagues24 found viral myelitis in 3 of 128 AIDS patients with neurologic symptoms, and the collective incidence of viral myelitis in AIDS was 1% when data were pooled from three different hospital series of neurologic complications of AIDS.27 The most common form of myelopathy observed with HIV infection has been a unique degeneration of the spinal cord first described by Petito and colleagues.28 In pathologic series, spinal cord degeneration has been observed in 11%29 to 22%28 of unselected cases. These pathologic series suggest that spinal cord disease occurring in association with HIV infection is common but clinically underrecognized.
The prototypic myelopathy observed with HIV infection is a unique degeneration of the spinal cord.28,30 Petito and colleagues28 observed this myelopathy in 20 of 89 consecutive autopsies of AIDS patients. Although the clinical presentation of this myelopathy may overlap with the presentation of other myelopathies associated with HIV-1 infection, the pathologic appearance is quite distinct. Clinically, patients complain of leg weakness, unsteadiness, and gait impairment. Incontinence of bladder and bowel often supervenes. In one study, incontinence was observed in 60% of patients.28 Patients with this disorder often complain of paresthesias and vague discomfort in their legs. Frequently, these complaints are attributed to the general debilitation of the patient, and the true nature of the illness remains undiagnosed until pathologic examination of the spinal cord at the time of autopsy.
On physical examination, a spastic paraparesis is detected with the degree of weakness exceeding the degree of spasticity. Rarely, marked asymmetry of the leg weakness, a monoparesis, or quadriparesis may be found. Gait ataxia is seen, and the heel-to-knee-to-toe test may reveal dysmetria and dyssynergy. Occasionally, weakness is slight or absent on confrontation testing, but hyperreflexia of the lower extremities and extensor plantar responses are noted. Muscle stretch reflexes may also be diminished or absent in this disorder, however, as a result of concomitant peripheral neuropathy. Sensory examination reveals that vibratory and position sense are disproportionately affected compared with pinprick, temperature, or light touch. A significant impairment of the latter modalities suggests the presence of a concomitant peripheral neuropathy. Electrophysiologic studies may reveal a prolonged latency of cortical evoked responses after tibial nerve stimulation. Typically, this myelopathy is seen late in the course of HIV-1 infection; however, it has been described as the presenting manifestation of infection.31
Gross examination of the spinal cord and dura is generally normal in HIV-associated myelopathy except when the myelopathy is particularly severe.28 The striking finding on histologic examination is the loss of myelin and spongy degeneration. The lateral and posterior columns are more severely affected than the anterior columns. Microvacuolization of the white matter of the spinal cord (Fig. 37–1) associated with lipid-laden macrophages bears an uncanny resemblance to the pathology of subacute combined degeneration of the spinal cord. The vacuolization seems to result from intramyelin swelling. Axons are preserved except in areas of marked vacuolization. Microglial nodules may be detected in the spinal cord gray matter, and 20% of the patients in one series also exhibited central chromatolysis of the anterior horn motor cells.28 Inflammation and intranuclear viral inclusions are generally not seen.
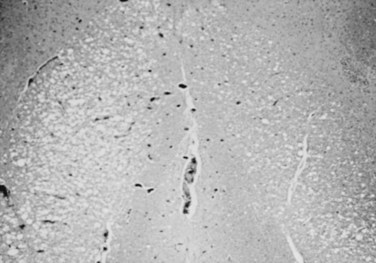
FIGURE 37–1 Microvacuolization of posterior columns of thoracic spinal cord in a patient with HIV-1–related myelopathy.
Although HIV has been cultured from the spinal cord, the specific role of HIV in the causation of this illness is uncertain. A similar clinicopathologic condition has been observed in patients with cancer or other immunosuppressive conditions in the absence of HIV infection.32 The spinal cord pathology is most prominent in the middle and lower thoracic regions. The cord involvement may be asymmetrical and does not seem to be confined to particular tracts.28 Goldstick and colleagues30 described involvement of the posterior columns increasing in intensity with rostral progression, whereas pyramidal tract involvement increased caudally. Petito and colleagues28 were able to correlate the frequency and severity of symptoms to the degree of spinal cord pathology in their series. A potential role of nutritional deficiency as the cause of spinal cord pathology has been suggested.33 Vacuolar myelopathy is not observed in young children with AIDS; however, pathologic abnormalities of the spinal cord are frequently seen at autopsy.34 A loss of myelin and axons in the corticospinal tracts seems to be the most common abnormality.
The diagnosis of this illness is one of exclusion. Numerous other myelopathies have been observed in association with HIV infection. An acute myelopathy of uncertain pathogenesis has been noted at the time of seroconversion.35 Table 37–3 lists other etiologies of spinal cord disease occurring in association with HIV.
TABLE 37–3 Potential Etiologies of the Myelopathies Associated with Human Immunodeficiency Virus (HIV) Infection
| Infectious |
| Viral |
| Bacterial |
| Fungal |
| Parasites |
| Noninfectious Etiologies |
Human T-Cell Lymphotropic Virus Type I
Although thought to be rare in the United States, human T-cell lymphotropic virus type I (HTLV-I) has been observed with increasing frequency in certain subpopulations. A study of volunteer blood donors by the American Red Cross revealed a seropositivity rate of 0.025% among volunteer blood donors.36 Intravenous drug abusers seem to be at particularly high risk for infection with HTLV-I. Seroprevalence rates in this population range from 7% to 49%.37,38 In a study of the seroprevalence among female prostitutes in eight areas of the United States, 6.7% were seropositive for HTLV-I and HTLV-II with prevalence rates ranging from 0% in southern Nevada to 25.4% in Newark, New Jersey.39 A case of transmission of HTLV-I by blood transfusion associated with myelopathy has been confirmed.40 In 10 native-born cases in the United States, 5 had received blood transfusions, and 6 had multiple sex partners (including one who frequented prostitution and had a history of drug abuse).41
In addition to being associated with adult T-cell leukemia/lymphoma, HTLV-I is associated with a chronic progressive myelopathy.42,43 An association between the presence of seropositivity for HTLV-I and multiple sclerosis has also been suggested,44 but this remains controversial.
The myelopathy that occurs with HTLV-I has been referred to as tropical spastic paraparesis (TSP) or HTLV-I associated myelopathy (HAM). This myelopathy is characterized on neuropathologic studies by chronic involvement of the pyramidal tracts chiefly at the thoracic level resulting in spastic lower extremity weakness and a spastic bladder. Paresthesias, pain, and sensory disturbances may also be observed. It is estimated that 1 in 250 individuals infected with HTLV-I develop this progressive myelopathy.45 The major pathologic features of HTLV-I myelopathy are long tract degeneration and demyelination affecting the pyramidal, spinocerebellar, and spinothalamic tracts associated with hyalinoid thickening of the media and adventitia of blood vessels in the brain, spinal cord, and subarachnoid space with perivascular cuffing with leukocytes, astrocytic gliosis, and foamy macrophages.46 These lesions may extend from the upper cervical cord to the lumbar regions. Vacuolization may be observed at the periphery of the lesions.
HTLV-II, a related type C retrovirus in the Oncoviridae subfamily also rarely may result in a myelopathy similar to HAM or TSP.47–53 The epidemiology of this virus is different from HTLV-I because the populations principally affected are American Indians and intravenous drug abusers.54 The mode of transmission parallels that of HTLV-I and HIV. The rarity of these neurologic disorders accompanying HTLV-II has precluded meaningful analysis of treatment options. The same therapies employed in HAM or TSP have been generally recommended, however.
Herpesviruses
Varicella-zoster virus (VZV) is responsible for varicella (chickenpox) and causes shingles in adults. The virus remains latent within the dorsal root ganglia and spreads centrifugally along the corresponding nerves after reactivation resulting in a severely painful, blistering dermatomal eruption. Rarely, when the thoracic dermatomes are involved, the virus may spread centripetally and result in a necrotizing myelopathy.55 The myelitis complicating VZV infection in immunocompetent individuals typically occurs 1 to 2 weeks after the appearance of the dermatomal rash, although VZV myelitis may develop in the absence of a rash. The myelitis occurs at the level of the affected dermatome and results in paraparesis. In immunosuppressed hosts, such as AIDS patients, VZV myelopathy occurs insidiously and progresses. The disease is suspected by the close temporal relationship to the rash and is confirmed by showing the presence of VZV DNA by polymerase chain reaction in the CSF or VZV antibody in the CSF. Treatment with intravenous acyclovir (10 mg/kg every 8 hours) should be initiated.
Rarely, and usually with primary infection, HSV-2, the etiology of genital herpes, may cause a sacral radiculitis56 or an ascending myelitis.57 These neurologic complications are rarely observed with recurrent HSV-2. Epstein-Barr virus,58,59 the etiologic agent of infectious mononucleosis, and cytomegalovirus60 may also result in a transverse myelitis at the time of primary infection. In an immunocompromised host, HSV-1, HSV-2, and cytomegalovirus may result in myelitis.
Other Viruses
Many other viruses have been associated with transverse myelitis (see Table 37–2). Of patients with transverse myelitis, 20% to 40% have evidence of preceding or concurrent viral infection.2,5,7,61,62 Prompted by the increasing availability and efficacy of antiviral therapy and improved diagnostic techniques, in particular, polymerase chain reaction, these percentages are likely to increase.
Myelopathies Resulting from Bacterial Disease
Syphilis
Syphilis may affect the spinal cord in various ways.25 The pathology may be predominantly meningovascular or parenchymatous in nature. Gummas may grow within the substance of the cord or compress the cord by growth from the surrounding meninges. The clinical picture of spinal cord compression in syphilis may also arise as a result of hypertrophic pachymeningitis or vertebral lesions secondary to syphilitic osteitis. Table 37–4 presents a classification based on pathology and modified after the one proposed by Adams and Merritt.63
TABLE 37–4 Syphilis of the Spinal Cord
Adapted from Adams R, Merritt H: Meningeal and vascular diseases of the spinal cord. Medicine 23:181, 1944.
Tabes Dorsalis
Tabes dorsalis is the prototypic spinal cord disorder associated with syphilis. It is characterized by incoordination, pains, anesthesia, and various visceral trophic abnormalities.64 The earliest recognized descriptions of this disorder date to the mid-18th century. By the turn of the 20th century, tabes dorsalis was recognized with increased frequency and, according to Erb, was unequaled in frequency or importance by any other chronic disease of the spinal cord.
The pathology of tabes dorsalis is characterized by changes in the posterior spinal roots and posterior spinal columns (Fig. 37–2). Shrinkage of the spinal cord may be apparent by gross inspection. Leptomeningitis is evidenced by round cell infiltration. The dorsal columns are demyelinated, particularly in the regions of the fasciculus gracilis, root entry zone, and Lissauer tract. Astrocytic proliferation in the posterior columns is accompanied by an increase in connective tissue and thickening of the blood vessel walls. The lower spinal cord bears the brunt of the damage. Nerve fibers in the posterior root are destroyed and replaced by fibrosis. Frequently, lesions are observed in the anterior horns, cranial nerves, and brainstem.
Syphilitic Meningomyelitis
Syphilitic meningomyelitis occurs most commonly in individuals 25 to 40 years old, although it may arise in younger and older age groups. In most series, men predominate. The latency from the onset of the infection to the onset of symptoms with syphilitic meningomyelitis varies from 1 to 30 years with most arising within 6 years of infection.65
Other Forms of Spinal Syphilis
Hypertrophic pachymeningitis66 is an insidious, slowly progressive syphilitic process that results in spinal root and spinal cord dysfunction. A rare but well-recognized complication of tertiary syphilis is a gumma of the spinal cord. The clinical features of a spinal gumma are indistinguishable from an intramedullary glioma if it arises within the cord or may simulate the appearance of an extramedullary tumor if it arises from the meninges and compresses the spinal cord. Spinal cord infarction is a well-recognized complication of syphilis. Another vascular complication of syphilis is aortitis, which may eventuate in an aortic aneurysm resulting rarely in myelopathy by erosion of the vertebrae and ultimately compression of the spinal cord. Progressive spinal muscular atrophy has been observed in association with neurosyphilis, although the relationship may be coincidental. Occasionally, syphilitic caries may affect the vertebrae, particularly vertebrae of the cervical spine, resulting in pain, tenderness to palpation, and loss of mobility, and an abnormal spinal curvature in the involved area is noted. Radicular pains are observed in one third of patients. Compression of the spinal cord, although rare, has been reported.
As with other forms of neurosyphilis, the recommended treatment is 12 to 24 million U of aqueous penicillin daily in divided doses administered every 4 hours for 10 to 14 days.67 Other treatment regimens68 using doxycycline, ceftriaxone, or erythromycin may be considered if the patient cannot tolerate penicillin. These treatment regimens are not well established in treating symptomatic neurosyphilis, however.
Tuberculosis
Neurologic complications of Mycobacterium tuberculosis remain common in some parts of the world but are rare in the developed countries of the Western world. Myelopathy occurring in association with tuberculosis is usually the consequence of tuberculous spondylitis (Pott disease), which accounts for half of all skeletal tuberculosis. Typically, the anterior portion of the vertebral body is affected, with the mycobacterial spread to the vertebrae being hematogenous, lymphatic, or by direct contiguity from the lung.69 The characteristic radiographic defect is anterior wedging of two adjacent vertebrae with loss of the intervening disk space. The spine is enveloped by pus extruding anteriorly from the affected vertebrae. Myelopathy typically results from pressure on the anterior spinal cord by caseous or granulating tissue, inflammatory thrombosis of the anterior spinal artery, or injury to the cord from spinal instability. The last-mentioned may lead to complete spinal cord transection.
Myelopathy occurring in association with tuberculous infection may also occur as a consequence of intraspinal granulomatous tissue unassociated with a bony lesion and intramedullary tuberculomas. In one study of spinal tuberculosis,70 54% of patients had neurologic deficit with bony tuberculous lesions, 39% had neurologic deficit with intraspinal granulomatous tissue occurring in the absence of bony lesions, and 7% had neurologic deficit from intraspinal tuberculomas.
Therapy of patients with spinal tuberculosis requires at least 12 months of antibiotic treatment and surgical decompression in the presence of neurologic abnormalities.70 In the setting of intraspinal granulomatous disease without significant bony destruction, laminectomy and débridement is adequate70; however, more aggressive therapy is warranted when vertebral bodies are involved. A two-stage procedure comprising posterior instrumental stabilization followed by anterior radical decompression permitted earlier mobilization after neurologic recovery.71 Reports of the frequency of neurologic recovery with spinal tuberculosis vary, but functional recovery rates of 90% have been reported.71 Thoracic lesions with severe neurologic deficit show the least improvement, whereas lumbar disease has the best outcome.72
Other Forms of Bacterial Myelopathy
Numerous other bacterial infections have been associated with myelitis. Rarely, the spinal cord may be seeded by bacteria leading to a suppurative myelitis with abscess formation. In a review by Dutton and Alexander,73 direct spread for adjacent infections was most commonly observed; however, hematogenous dissemination from endocarditis, pulmonary infections, and other sites was also frequently observed. Organisms isolated in these cases have included staphylococci, streptococci, Escherichia coli, and Nocardia. Rarely, Whipple disease can manifest as a myelopathy.74,75
More often, myelopathies associated with bacterial infection are parainfectious in nature, similar clinicopathologically to myelopathies occurring after viral infection or vaccination. Potential causes76,77 include scarlet fever, pertussis, whooping cough, mycoplasmal pneumonia, and pneumococcal pneumonia. Myelitis resulting in Brown-Séquard syndrome has also been described with cat-scratch disease.78 Lyme disease, the result of infection with Borrelia burgdorferi, a treponeme, may also result in myelopathy.79

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


