Chapter 15 Mechanisms of Acute Inflammation and Vascular Injury in SLE
Epidemiology of Premature Vascular Damage in SLE
Accelerated atherosclerosis is an important problem in patients with SLE (see Chapter 26 for more detail). Enhanced atherosclerotic risk increases with each year of disease duration. This is especially the case in young females with SLE, in whom the cardiovascular risk can be up to 50-fold higher than in age-matched controls (Figure 15-1).1,2 Clinically evident coronary artery disease affects approximately 6% to 10% of patients with SLE.1 There is also evidence that SLE, like diabetes mellitus, increases risk of poor outcomes after acute myocardial infarction.3
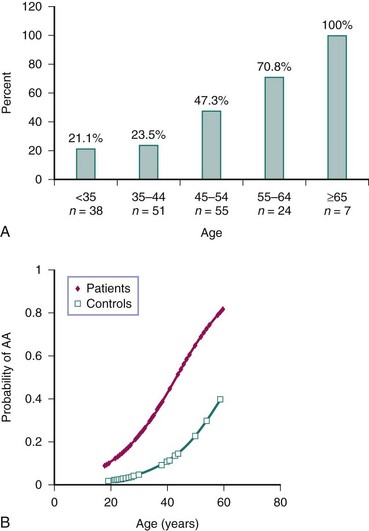
FIGURE 15-1 Cardiovascular disease is increased in SLE.
(A from Manzi S, et al: Prevalence and risk factors of carotid plaque in women with systemic lupus erythematosus. Arthritis Rheum 42:51–60, 1999; B from Roldan C, et al: Premature aortic atherosclerosis in systemic lupus erythematosus: a controlled transesophageal echocardiographic study. J Rheumatol 37:71–78, 2010.)
Traditional Framingham Study risk factors likely contribute to CVD in SLE, but they cannot fully account for the increased risk. Therefore, the pathogenesis of premature CVD in SLE may also rely on factors unique to the disease itself.4 Many investigators agree with the hypothesis that long-term exposure to lupus immune dysregulation promotes CVD.
The type of cardiovascular lesion may provide clues to its etiology. The pathology of SLE-related CVD can involve atherosclerotic lesions5 and also fibrointimal hyperplasia, which may reflect chronic endothelial injury (Figure 15-2).6 Noncalcified coronary plaque is more common in patients with currently active or recently active SLE, suggesting that disease-specific factors directly contribute to development of new plaque.7 Contrarily, calcified plaque correlates with traditional cardiovascular risk factors in patients with lupus, suggesting that factors such as age and obesity may advance calcified disease.8
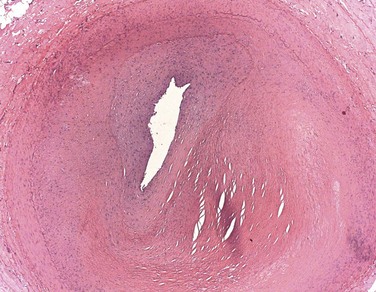
FIGURE 15-2 Hematoxylin and eosin stain of a coronary artery from a 35-year-old woman with lupus.
(Photomicrograph courtesy of Dr. Gerald Abrams.)
Subclinical and Clinical Vascular Damage in SLE
Premature damage in SLE has been described in both the macrovasculature and microvasculature. Vascular functional abnormalities in lupus appear to develop early during the course of the disease,9 although it is unclear whether they precede SLE diagnosis. Subclinical vascular dysfunction can be quantified with a variety of invasive and noninvasive tests (see Chapter 26); patients with SLE have significantly decreased flow-mediated dilation (FMD) of the brachial artery—a function of endothelial cells—and this decrease correlates with increased carotid intima media thickness (IMT) in such patients.10 Additionally, carotid plaque can be detected in 21% of patients with SLE younger than 35 years and in up to 100% of those older than 65.11 Aortic atherosclerosis is also increased in SLE.12 Subclinical macrovascular disease in SLE correlates with disease activity and disease duration.10–12 Damage to the coronary circulation is also common in patients with SLE; in one study, more than half of the tested patients displayed noncalcified coronary plaque.7 There is also evidence of impairment of the coronary microvasculature flow reserve and altered coronary vasomotor function, even in those patients with lupus who have grossly normal coronary arteries.13 This dysfunction correlates with disease duration and severity, suggesting that microvascular damage and dysfunction are also part of SLE-related cardiovascular pathology.14
Mechanisms of Atherosclerosis Development in the General Population
Inflammation is considered to play a crucial role in the pathogenesis of atherosclerosis and is present throughout the various stages of the vascular damage process. In early lesions of atherosclerosis, the fatty streak and infiltration by macrophages and T cells are prominent. Indeed, factors such as oxidized LDL (ox-LDL) activate the endothelium to secrete chemokines that recruit inflammatory cells, including T lymphocytes, dendritic cells (DCs), and monocytes. The monocytes differentiate into macrophages and foam cells under the influence of locally secreted factors and further stimulation by ox-LDL.15,16 Cholesterol crystals and other stimuli activate macrophages and foam cells to secrete inflammatory cytokines, reactive oxygen and nitrogen species, and proteases. All these factors can contribute to the atherogenic phenotype in the blood vessel.17 Invasion of the atherosclerotic plaque by CD4+ T cells also contributes to vascular pathology, through these cells’ recognition of epitopes of various molecules, such as ox-LDL, and secretion of interferon gamma (IFN-γ), which then leads to increased inflammatory cytokine production. Chronic and unabated synthesis of proteases and inflammatory cytokines promotes thinning of the atherosclerotic plaque wall and eventual rupture. Rupture leads to exposure of the blood to phospholipids, tissue factor, and platelet-adhesive matrix molecules, eventually promoting thrombosis and acute cardiovascular events.15
Under normal conditions, vascular damage triggers a response that leads to an attempt to repair the endothelium. If this repair fails or is incomplete, the vessel may be at a higher risk for atherosclerotic disease. Repair of the damaged endothelium has been proposed to occur primarily by circulating bone marrow–derived endothelial progenitor cells (EPCs) and by myelomonocytic circulating angiogenic cells (CACs).18 Decreased numbers or dysfunction of these cell types may contribute to CVD in persons with various diseases as well as in the general population, because EPC numbers inversely correlate with CVD risk, time to first cardiovascular event, and in-stent re-stenosis risk.19,20 Additionally, functional impairment of EPCs correlates with coronary artery disease risk.21
Mechanisms of Endothelial Inflammation, Injury, and Atherosclerosis in SLE
Because of the profound immune dysregulation present in SLE, the increased risk of CVD is likely secondary to a combination of many factors that alter the endothelium and inflammatory response. Indeed, variables that enhance traditional mechanisms of atherosclerosis and create novel injury pathways are present in this disease (Box 15-1). As such, understanding the putative mechanisms that promote accelerated vascular damage in SLE may also provide novel information about the pathways by which the immune system may promote atherosclerosis in the general population.
Box 15-1
Mechanisms of Vascular injury in SLE
1. Increased endothelial damage:
3. Enhanced plaque formation by IFN-α.
4. Neutrophil extracellular traps induce endothelial cell death.
5. IFN-α–activated platelets promote vascular inflammation.
6. Dysregulated cytokine production:
7. CD154-CD40 interactions and CD137 co-stimulation lead to vascular damage by T cells.
9. Autoantibodies with various targets affecting many steps in the atherogenic cycle.
Endothelial damage is increased in SLE. Patients with SLE have increased numbers of circulating apoptotic endothelial cells, which correlate with endothelial dysfunction (as assessed by brachial artery FMD) and circulating levels of tissue factor (Figure 15-3A).9 Various soluble adhesion molecules, such as vascular cell adhesion molecule (VCAM), intercellular adhesion molecule (ICAM), and E-selectin, which are released after endothelial cell damage, are increased in SLE and correlate with higher coronary calcium scores.22 Additionally, soluble levels of the antithrombotic endothelial protein C receptor, which is typically released secondary to inflammatory activation of metalloproteinases, are increased in SLE and correlate with the presence of carotid plaque.23 These findings indicate that the endothelium is under chronic assault in SLE, a phenomenon that could lead to atherosclerotic pathology if the damage is not adequately repaired.
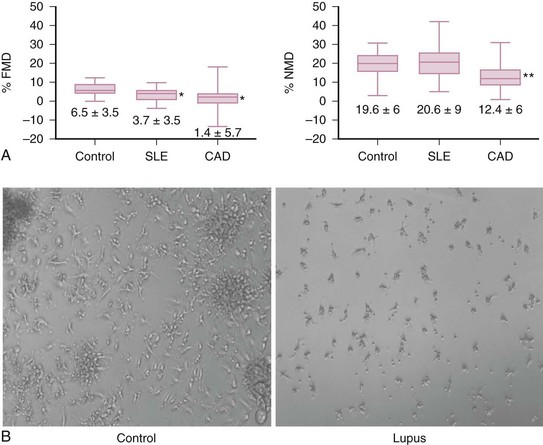
FIGURE 15-3 A, Endothelial dysfunction is characteristic of SLE-mediated cardiovascular disease.
B, Vascular repair is dysfunctional in SLE.
(A from Rajagopalan S, et al: Endothelial cell apoptosis in systemic lupus erythematosus: a common pathway for abnormal vascular function and thrombosis propensity. Blood 103:3677-3683, 2004.)
However, despite evidence that accelerated endothelial cell death occurs in lupus, a phenomenon that should trigger enhanced vascular repair, the latter is significantly impaired in SLE. Patients with lupus, even those with very stable disease, have decreased circulating EPCs. Further, EPCs/CACs in SLE exhibit enhanced apoptosis and demonstrate decreased capacity to synthesize proangiogenic molecules, to be incorporated into vascular structures, and to differentiate into mature endothelial cells (Figure 15-3B).24–26 Thus, patients with SLE have compromised repair of the damaged endothelium, and we can hypothesize that this phenomenon may contribute to the establishment of a milieu that promotes the development of vascular plaque.
Type I Interferons and SLE-Related Cardiovascular Disease
One mechanism by which vascular repair is impaired in SLE is through increased levels and enhanced effects of type I IFNs, cytokines known to play important roles in innate immunity and antiviral responses. Human and murine studies from various groups indicate that IFN-α may be crucial in the pathogenesis of SLE. Approximately 60% of patients with SLE have elevations of serum IFN-α and carry an “IFN signature” in peripheral blood mononuclear cells, kidneys, and other tissues, in correlation with disease activity.27,28 Further, lupus cells appear to be more sensitive to the effects of type I IFNs.29 Because of the role of type I IFNs in SLE pathology, these cytokines have been investigated as a contributing factor to the development of lupus-related CVD. A summary of the mechanisms by which IFN-α insults the vasculature is shown in Figure 15-4.
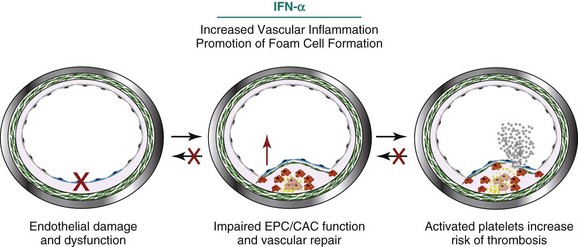
FIGURE 15-4 Interferon-α (IFN-α) contributes to SLE-mediated vascular disease in a variety of ways.
(Artwork partially contributed by Seth G Thacker.)
Over the past few years, evidence has surfaced that type I IFNs correlate with atherosclerosis and endothelial dysfunction. Patients with lupus and a high type I IFN signature have decreased endothelial function, as assessed by peripheral arterial tone measurements.30 Additionally, type I IFN serum activity in SLE was found to be positively associated with increased carotid IMT, coronary calcification, and decreased brachial artery FMD in a cohort of patients with lupus who had low traditional cardiovascular risk factors, stable disease, and no previous cardiovascular events.31 Importantly, in this cohort, factors such as high-sensitivity CRP, serum levels of adhesion molecules, and lupus disease activity were not associated with functional or anatomic evidence of vascular damage in SLE. Thus, enhanced type I IFN effects may be one of the unique factors in SLE that results in increased cardiovascular risk.
Induction of an Imbalance of Vascular Damage and Repair by Type I Interferons
Type I IFNs promote decreased vascular repair. In SLE, dysfunction of EPC/CAC differentiation is mediated by IFN-α, because neutralization of this cytokine restores a normal phenotype of these cells.26 This theory is further reinforced by the observation of abrogated EPC/CAC numbers and function in lupus-prone New Zealand Black/New Zealand White F1 mice, a strain that depends on type I IFNs for disease development and severity. Additionally, non–lupus-prone mouse EPCs are unable to properly differentiate into mature endothelial cells in the presence of IFN-α.32,33 The pathways by which IFN-α mediates aberrant vascular repair may depend on repression of the proangiogenic factors interleukin-1β (IL-1β) and vascular endothelial growth factor (VEGF) and on upregulation of the cytokine IL-18 and the antiangiogenic IL-1 receptor antagonist. Indeed, addition of recombinant human IL-1β to SLE EPC/CAC cultures restores normal endothelial differentiation.32,34 Further, blockade of IL-18 signaling improves endothelial differentiation, suggesting that the balance between IL-1β and IL-18, which is modulated by IFN-α, may be important in vascular health.34 There is evidence that an antiangiogenic phenotype is operational in vivo in patients with SLE, as manifested by decreased vascular density and increased vascular rarefaction in renal blood vessels, in association with upregulation of the IL-1 receptor antagonist and decreased vascular endothelial growth factor in the kidney and serum.26,32
The cellular source of type I IFNs leading to abnormal vascular repair has been examined. Depletion of plasmacytoid DCs (pDCs; the major in vivo producers of IFN-α) does not lead to abrogation of abnormal lupus EPC/CAC differentiation in culture.35 Therefore, other cellular sources for this cytokine in the context of interactions with the endothelium have been sought. Neutrophil-specific genes are abundant in peripheral blood mononuclear cell microarrays from patients with lupus because of the presence of low-density granulocytes (LDGs) in mononuclear cell fractions.18,36 These LDGs have the capacity to secrete sufficient amounts of IFN-α to interfere with vascular repair. Indeed, LDG depletion from lupus peripheral blood mononuclear cells restores the ability of EPC/CACs to differentiate in vitro into endothelial monolayers.35
IFN-α and Plaque Formation
In addition to the role type I IFNs play in modulating endothelial cell death and repair, they may also contribute to plaque development through other mechanisms. For example, IFN-α–producing plasmacytoid DCs have been identified in areas of atheromatous plaque from patients without SLE. IFN-α then activates plaque-residing CD4+ T cells to increase expression of tumor necrosis factor (TNF)–related apoptosis-inducing ligand (TRAIL), which results in killing of plaque-stabilizing cells and a potential increase in the risk of plaque rupture. Additionally, IFN-α sensitizes plaque-residing myeloid DCs, potentially promoting further inflammation and plaque destabilization. This cytokine can act in conjunction with bacterial products (such as lipopolysaccharide [LPS]) to increase the synthesis of various proinflammatory cytokines and metalloproteinases.32,37,38 Importantly, IFN-α has also been shown to upregulate the macrophage scavenger receptor A (SRA), which allows for ox-LDL uptake and foam cell formation. Indeed, SRA and CD36 have been implicated in foam cell formation and in the regulation of inflammatory signaling pathways leading to lesional macrophage apoptosis and plaque necrosis in other conditions. The presence of a strong interferon gene signature correlates with higher levels of macrophage SRA messenger RNA (mRNA) in patients with SLE, indicating an additional mechanism by which type I IFNs may modulate deleterious vascular responses in SLE.39 These findings indicate that type I IFNs could potentially be involved in atherosclerosis development not only in persons with autoimmune disorders but also in the general population in the context of idiopathic atherosclerosis and microbial infections.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


