Chapter 32 Management of Sjögren Syndrome in Patients with SLE
Sjögren syndrome (SS) is an autoimmune inflammatory disorder of the exocrine glands, which particularly affects the lacrimal and salivary glands. Frequent symptoms are dry mouth and dry eyes, often in conjunction with several nonspecific symptoms, such as malaise and fatigue. In addition, extraglandular manifestations, such as purpura, polyneuropathy, arthritis, and others, can be presenting signs of the disease (Table 32-1). The estimated prevalence of SS in the general population is between 0.5% and 1.0%, which makes SS the most common systemic autoimmune disease after rheumatoid arthritis (RA). SS is more frequent in women than in men, with a female-to-male ratio of 9 : 1. SS can be a primary idiopathic condition of unknown cause (primary SS [pSS]), but it may also occur in the presence of another autoimmune disorder such as RA, systemic lupus erythematosus (SLE), scleroderma, or mixed connective tissue disease (secondary SS [sSS]). In RA, the prevalence of sSS is approximately 30%; in SLE, approximately 20% of patients fulfill the classification criteria for sSS. Further, SS is associated with organ-specific autoimmune diseases; in particular, autoimmune thyroid disease, primary biliary cirrhosis, and autoimmune gastritis, which underscores the autoimmune nature of the disease.1,2 Patients with SS may be restricted in their activities and participation in society, resulting in a reduced health-related quality of life and an impaired socioeconomic status.3
TABLE 32-1 Estimated Prevalence of a Particular Extraglandular Manifestation among Patients with Sjögren Syndrome7,96,97,101
| AFFECTED ORGAN SYSTEM | EXTRAGLANDULAR MANIFESTATIONS | ESTIMATED PREVALENCE (%)* |
|---|---|---|
| Joints and muscles | Arthralgia or arthritis | >50 |
| Myopathy | 22 | |
| Skin | Xerosis | >50 |
| Cutaneous vasculitis (purpura) | 10 | |
| Other skin lesion (e.g., erythema nodosum, livedo reticularis, lichen planus, vitiligo, cutaneous amyloidosis, and granuloma annulare) | <5 | |
| Raynaud phenomenon vasculitis | 13-30 | |
| Cardiovascular | Pericarditis | Up to 30 |
| Respiratory tract | Interstitial lung disease (generally mild) | 30 |
| Mucosa-associated lymphoid tissue (MALT) lymphoma | 5-9 | |
| Gastrointestinal tract | Dysphagia | >50 |
| Oesophageal involvement | 36-90 | |
| Gastritis | 20 | |
| Nervous system | Peripheral neuropathy | 20 |
| Cranial neuropathy | 5 | |
| Central nervous system (CNS) involvement (focal or generalized) | Up to 20 | |
| Urogenital tract | Interstitial nephritis with renal tubular acidosis | 25 |
| Glomerulonephritis (associated with cryoglobulinemia) | <10 | |
| Interstitial cystitis | 4 |
* Percentages greatly differ among studies.
History
The first descriptions of SS were reported by European clinicians between 1882 and 1925.4 In 1892, Mikulicz observed a man with bilateral parotid and lacrimal gland enlargement that was associated with massive round-cell infiltration. In 1925, Gougerot described three patients with salivary and mucous gland atrophy and insufficiency. In 1933, a Swedish ophthalmologist named Henrik Sjögren reported clinical and histologic findings in 19 women with xerostomia and keratoconjunctivitis sicca (KCS), of whom 13 had chronic arthritis. Seminal work by Morgan, Castleman, Bunim, and Talal in New York and at the National Institutes of Health in the 1950s and 1960s established Sjögren syndrome as an autoantibody-associated autoimmune disorder, detailed its clinical features, and recognized its association with lymphoma.5
Clinical Presentation
Glandular Manifestations
SS affects the exocrine glands, in particular, the lacrimal and salivary glands, resulting in sicca complaints, or dry eyes, and a dryness of the oral cavity. With respect to the eyes, symptoms of burning, sandy sensations with pain, and photophobia and photosensitivity prevail (Table 32-2). Physical examination reveals chronic irritation and destruction of both corneal and bulbar conjunctival epithelial KCS as a result of disturbed tear production. Accumulation of thick, ropelike secretions along the inner canthus may be the result of decreased tear film and an abnormal mucous component. Progressive keratitis may result in a loss of vision. A common problem in patients with dry eye is blepharitis. Conjunctivitis, as a result of secondary infection with Staphylococcus aureus, may also occur.
TABLE 32-2 Onset and Duration of Symptoms of Eye and Mouth Dryness in Patients with and without Sjögren Syndrome98

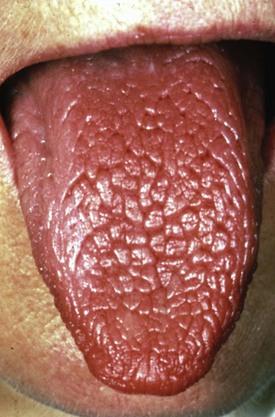
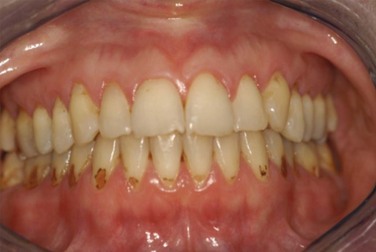
Reduced saliva production induces the sensation of dry mouth (xerostomia). Typical dryness-related complaints in early SS are predominantly present at rest and during the night. Over time and as SS develops, the dryness is also noted during the day.6 Physical examination shows a dry mouth and tongue with an adherent, sticky mucus that coats the erythematous mucosa. The tongue may be smooth and reddened with some loss of dorsal papillae, or it may have a fissured appearance (Figure 32-1). The lips often appear cracked, peeling, and atrophic and may even appear furrowed or pebbled. The buccal mucosa may be pale and corrugated in appearance. Dental caries is not uncommon (Figure 32-2), as well as secondary infection of the mucosa with Candida albicans. Enlargement of the salivary glands may be present and is, generally, due to the presence of an autoimmune inflammatory process. However, enlargement of the glands might also be the result of lymphoma development or secondary infection caused by stasis of saliva.
Extraglandular Manifestations
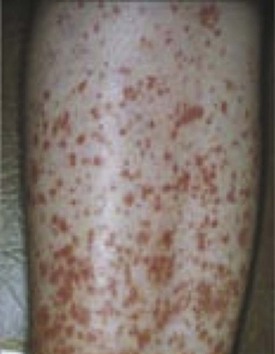
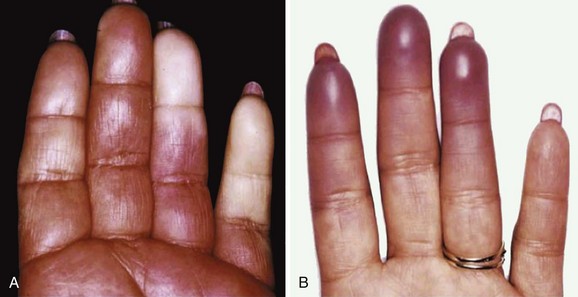
SS is a systemic autoimmune disease in which many different organs may be affected, giving rise to the various extraglandular clinical manifestations7 (see Table 32-1) (Figures 32-3 and 32-4). The involvement of extraglandular organs can go unrecognized until clinical symptoms become apparent in later stages of the disease. In addition, general systemic symptoms such as fatigue, myalgia, and depression are frequently present.
Lymphoma Development
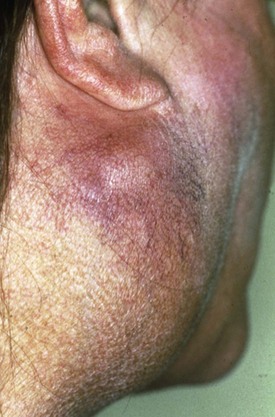
Lymphomas develop in approximately 7.5% of patients with SS. Moreover, patients with SS have an 18.8 (CI 9.5 to 37.3) times increased risk of developing lymphomas.8 In most cases these are marginal zone B-cell lymphomas occurring in the salivary glands, in particular the parotid gland, the so-called mucosa-associated lymphoid tissue (MALT) lymphoma. These lymphomas are generally localized and follow an indolent, rather benign, clinical course. In a minority of patients, aggressive non-Hodgkin lymphoma (NHL) is present and even Hodgkin disease has incidentally been described in SS. Risk factors for the development of lymphoma include the presence of cryoglobulins, low complement C4 levels, and palpable purpura.9,10 The presence of germinal center–like structures in salivary gland biopsies is highly predictive for the development of lymphoma.11 Isolated salivary gland enlargement, as well as any persistent lymph node swelling (Figure 32-5), in a patient with SS should raise the suspicion of lymphoma development.
Serologic Findings
The most characteristic autoantibodies in SS are anti–Sjögren syndrome antigen A (anti-SSA/Ro) antibodies, present in 70% of patients, and anti–Sjögren syndrome antigen B (anti-SSB/La) antibodies, present in approximately 50% of patients. High titers of these autoantibodies, in particular anti-SSB/La antibodies, are associated with extraglandular disease. Anti-SSB/La antibodies are considered to be the most specific serologic marker for SS, although they can also be found in 25% to 35% of patients with SLE or other autoimmune connective-tissue disorders and in approximately 5% of healthy individuals. Anti–alph-fodrin autoantibodies occur in approximately 30% of patients with SS and are considered specific for the disease. Autoantibodies to human muscarinic acetylcholine receptor 3 are present in 90% of patients with pSS and 71% of patients with sSS; however, they are not specific for SS, because they are also present in 65% and 68% of patients with RA and SLE, respectively.12 The rheumatoid factor is present in approximately 50% of patients but has a very low specificity for SS. Between 10% and 20% of patients with SS demonstrate mixed essential cryoglobulins. The presence of cryoglobulins is associated with vasculitic manifestations such as purpura, polyneuropathy/mononeuritis multiplex, and glomerulonephritis, and they constitute a risk factor for the development of lymphoma. Hypergammaglobulinemia, present in 40% of patients, reflects polyclonal B-lymphocyte activation, which is characteristic of SS.13 In addition, monoclonal gammopathy, reported in 22% of patients, demonstrates excessive clonal B-cell proliferation and is associated with the development of lymphoma.
Classification and Diagnosis of Sjögren Syndrome
Many classification criteria for SS have been suggested. At present, the revised American-European classification criteria for SS, which were proposed in 2002, are the most widely accepted and validated criteria (Box 32-1). These criteria combine subjective symptoms of dry eyes and dry mouth with the objective signs of KCS and xerostomia.14
Box 32-1
Revised International Classification Criteria and Revised Rules for Sjögren Syndrome Classification
I. Ocular symptoms—Require a positive response to at least one of the following questions:
II. Oral symptoms—Require a positive response to at least one of the following questions:
III. Ocular signs (i.e., objective evidence of ocular involvement)—Are defined as a positive result for a least one of the following tests:
IV. Histopathology—In minor salivary glands (obtained through normal-appearing mucosa), focal lymphocytic sialadenitis, evaluated by an expert histopathologist, with a focus score of ≥1, defined as a number of lymphocytic foci (adjacent to normal-appearing mucous acini and contain more than 50 lymphocytes) per 4 mm2 of glandular tissue
V. Salivary gland involvement—Objective evidence of salivary gland involvement is defined by a positive result to at least one of the following diagnostic tests:
VI. Autoantibodies—Presence in the serum of the following autoantibodies:
Revised Rules for Classification
For Primary Sjögren Syndrome
a. Presence of any four of the six items is indicative of pSS, as long as either item IV (histopathology) or VI (serology) is positive.
b. Presence of any three of the four objective criteria items (items III, IV, and VI) is indicative of pSS.
c. Classification tree procedure represents a valid alternative method for classification, although it should be more properly used in a clinical epidemiologic survey.
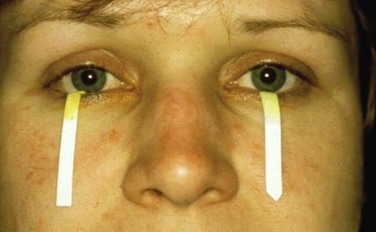
The subjective ocular and oral symptoms are obtained by history taking. Two tests are used to objectify reduced tear production. In the Schirmer test a piece of filter paper is placed laterally on the lower eyelid, which results in wetting due to tear production. If less than 5 mm of the paper is wetted after 5 minutes, then the test result is considered positive (Figure 32-6). In the rose bengal test, dye stains devitalized areas of the cornea and conjunctiva, which can then be scored using a split lamp. A rose bengal score of ≥4 according to the van Bijsterveld scoring system is considered abnormal. Instead of rose bengal stain, lissamine green can be used, which shows comparable results but is less painful. An additional test, which is not accepted as a diagnostic technique for SS but provides a global assessment of the function of the tear film, is the tear break-up time test. This test is performed by measuring break-up time after the instillation of fluorescein. An interval of less than 10 seconds is considered abnormal.
Currently, the most commonly applied noninvasive objective salivary gland diagnostic test is measuring the flow rate of unstimulated whole saliva. The patient is asked to expectorate once and then collect all saliva into a graduated container during a 15-minute period. Results obtained by sialometry, regardless of the presence of oral complaints, allow monitoring of the disease progression.6 If more specific functional information is required for a particular gland, for research purposes and for patient-based advice on how best to reduce xerostomia, then individual gland collection techniques can be used (Figures 32-7 and 32-8).
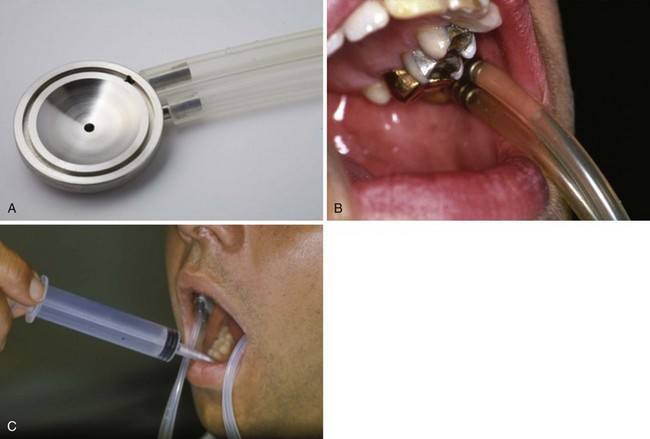
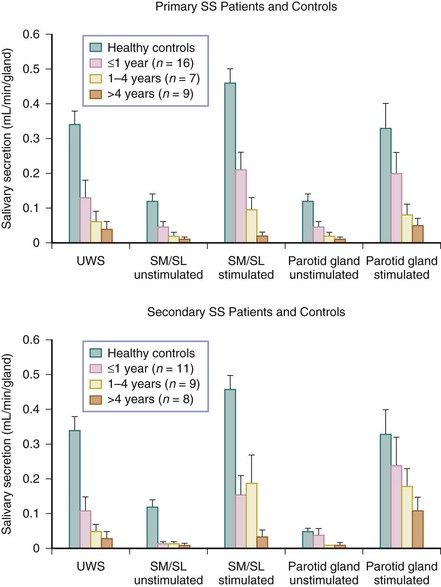
FIGURE 32-8 Relation among disease duration (i.e., the time from first complaints induced by or related to oral dryness until referral), mean salivary flow rates (mean ± SEM), unstimulated whole saliva (UWS), and submandibular/sublingual glands (SM/SL).16
To confirm the diagnosis of SS histopathologically, usually a biopsy from a labial salivary gland is taken. The diagnosis can be confirmed if this biopsy shows focal lymphocytic sialadenitis with a focus score, defined as an accumulation of 50 or more lymphocytes per 4 mm2, of ≥1.15 Recently, it has been shown that parotid biopsies might serve as a proper alternative in the diagnosis of SS. In such biopsies, MALT or NHL pathologic findings are easier to detect because parotid glands are more commonly affected,16 and the same gland can be biopsied more often.17 Imaging studies can also be used to evaluate salivary gland involvement. Sialography of the parotid gland has a high diagnostic accuracy. The main characteristic of SS is a diffuse collection of contrast fluid at the terminal acini of the ductal tree, called sialectasia.18,19 With scintigraphy, patients with SS demonstrate decreased uptake and release of technetium (Tc)–99m pertechnetate.20 Finally, submandibular ultrasound might be a promising, less-invasive alternative to sialography in the classification of SS.21
The presence of nonspecific serologic markers of autoimmunity such as antinuclear antibodies, rheumatoid factors, and elevated immunoglobulins (particularly IgG) are important contributors to a definitive diagnosis of SS.22
Pathogenesis
Most of what is known about the pathogenesis of SS comes from patients with pSS, whereas little specific information is available about the pathogenesis of sSS. Furthermore, much of the work cited in the following text relies on two experimental models of SS: (1) nonobese, diabetic (NOD) mice that are autoimmune prone and diabetic but independent of the usual diabetes and insulinitis in that strain, and (2) B cell–activating factor (BAFF) generated transgenic mice, in which enhanced survival is sufficient to induce disease. Unfortunately, both of these models lack the autoantibody profile (e.g., anti-SSA/Ro, anti-SSB/La) of the human counterpart.23
Genetic Factors
Human leukocyte antigen (HLA)–DR and HLA-deterodimer (HLA-DQ) class II genes are involved in the pathogenesis of SS but vary with geography and ethnicity.24 B8, DWw52, DR2, Gla3, and 5 probably each play a role. Anti-SSA/Ro 52 autoantibody production is linked to DRB1*0301, DRB3*0301, DQA1*0501, and DQB1*0201. Genetic-wide association studies have correlated innate immune interferon regulatory factor 5 (IR5) rs2004640 T allele (odds ratio 1.93) polymorphisms, as well as STAT4 (SNP rs7582694), which encodes transcription factors involved with interferon signaling with SS.
Hormonal Factors
SS is far more common among women than men. X-chromosome silencing and sex steroids probably play important roles in the pathogenesis of SS.25 Many genes on the X chromosome are involved in the immune system, and impaired inactivation of the genes on the X chromosome may contribute to the development of autoimmune disease. Sex hormones have key roles in the function of cells of the immune system. Estrogens appear to have positive effects on the emergence of autoimmune disease, and androgens have a more protective role, although the mechanisms are not well understood. Patients with SS appear to be androgen deficient and have lower serum concentrations of dehydroepiandrosterone (5-DHEA) and its sulfate ester, dehydroepiandrosterone-sulfate (DHEA-S).26–28 Sex hormones also influence saliva and tear production.
Glandular Infiltration
Epithelial cells in SS glandular tissues are not only targets for the disease, but these cells also exert important immunologic functions. Ductal epithelial cells show enhanced expression of CD40 and adhesion molecules, as well as increased production of lymphoid chemokines, cytokines (including proinflammatory cytokines), and B lymphocyte stimulator (BlyS) or BAFF.23 Epithelial cells also express Toll-like receptors (TLRs). Glandular viral infections can prompt epithelial cells to activate the innate immune system via TLRs.29 A high incidence of Epstein-Barr virus reactivation in SS has been reported, and this virus commonly infects salivary epithelial glands and T cells. Binding of viral ligands to TLR3 on ductal cells is believed to be a major source of type I interferons in glandular tissue of patients with SS. One of the important cytokines induced by type I interferons is BAFF. The upregulation of adhesion molecules and the production of chemokines and cytokines promote the migration of lymphocytes and dendritic cells into epithelial glandular tissues. Epithelial cells derived from patients with SS are probably intrinsically activated. The infiltrating T cells and dendritic cells secreting proinflammatory cytokines, such as interleukin-1 beta (IL-1β), interferon-gamma (IFN-γ), and tumor necrosis factor alpha (TNF-α), promote further activation of the epithelial cells and inflammation of the glands. Through apoptosis and exosomes, epithelial cells present intracellular autoantigens, and anti-SSA/Ro and anti-SSB/La are translocated from apoptotic blebs where they trigger the production of autoantibodies against these antigens by local (infiltrated) B cells. Type I interferon-induced BAFF, produced by the epithelial cells, may well play a role in this B-cell activation. Not only BAFF, but also levels of a proliferation-inducing ligand (APRIL) are increased in patients with pSS. These two cytokines share receptors and play different but essential roles in the regulation of B-cell survival, differentiation, and proliferation. The elevated levels of these cytokines might be, at least partly, responsible for the presence of autoantibodies, hypergammaglobulinemia, oligoclonal B-cell expansion, ectopic germinal center–like structures, and increased risk for developing NHL.
Sjögren Syndrome in Patients with Lupus
The association of SS and SLE was first noted in 1959. Small-scale studies suggested the prevalence of SS in SLE ranging from 7% to 35%. Four recent, large-scale studies have analyzed the influence of SS on SLE. In one study, 9.2% of 283 patients with SLE who were Greek met the American-European classification criteria for SS.30 Patients with SLE who had SS tended to be older, have a higher prevalence of Raynaud phenomenon, rheumatoid factor, anti-SSA and anti-SSB, and a high frequency for the DRB1*0301 allele. These patients had less renal disease, adenopathy, and thrombocytopenia. Of the patients with SLE who were Norwegian, 81 patients over the age of 70 years were compared with matched individuals with RA and healthy control participants.31 The SLE group had more fatigue, anti-SSA and anti-SSB antibodies, and a positive Schirmer test. In the Johns Hopkins study, 259 (14%) of the 1531 patients with SLE were found to have SS by clinical evaluation.32 These patients were generally older, white women with more photosensitivity; oral ulcers; Raynaud phenomenon; less renal disease; and anti-SSA and anti-SSB, anti–double-stranded DNA (anti-dsDNA), and anti-ribonucleoprotein (anti-RNP) antibodies. Approximately 20% of more than 2000 patients with lupus who were of Chinese descent at Peking Union Medical College also had SS.33 Significant differences between those with SS/SLE and those with SLE only included older age, female gender, and higher rates of sicca symptoms and signs, renal tubular acidosis, and interstitial lung disease in the former. Patients with SLE only had more rash, nephrosis, central nervous system disease, lower IgG levels, more disease activity, and more immune suppressive and corticosteroid use. Seventy-one percent of the patients with SS/SLE were SSA or SSB positive versus 20% of those with SLE. In summary, patients with SS/SLE who met SS criteria made up approximately 10% of the SLE population, although twice that many have features of SS. These patients tend to be older and, Caucasian, to have a more benign process, and to less frequently require aggressive management.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


