Chapter 56 Investigational Agents and Future Therapy for SLE
When Moriz Kaposi1 in 1872 dealt with the therapy of systemic lupus erythematosus (SLE) for the first time in the history of medicine, bed rest, ointments, and plant extracts were the only available remedies for a disease whose cause and pathogenesis were unknown. When Philip Hench2 introduced glucocorticoids into antirheumatic therapy, their efficacy soon became apparent, and they were then successfully used for SLE—until today.3 However, pathogenetic insights were still not available and even the prototypic autoantibodies had not yet been characterized. In contrast, in more recent times, the view on SLE became enlightened by an understanding of the final autoantibody- and immune complex–mediated events; and, likewise, by the following developments: (1) the detection of a plethora of cytokines, chemokines, and similar mediators, including the possibly important role of type I interferons4; (2) the detection of apoptosis and its regulation5; (3) the definition of new cell populations that may be importantly involved; (4) the elucidation of a myriad of signal transduction pathways and transcription factors that regulate gene expression6; and (5) the description and sequence determination of SLE susceptibility genes.7
As the cause of SLE remains enigmatic and the disease is still incurable in many patients and associated with significant mortality,8 the insights from basic research activities pave the paths to new therapies. Indeed, although no new drugs for SLE have been licensed for many decades, very recently, belimumab, a biologic agent targeting (no surprise) B cells, has been approved for the treatment of refractory mild and moderate SLE. Many other approaches are still theoretical and will need subtle realization, but others are already in experimental and even in early clinical investigation. In this context, it is important to bear in mind that the life-threatening nature of severe SLE may not allow for traditional blinded controlled clinical trials early in the development of a new therapeutic regimen, but that ultimate proof of efficacy must still be achieved by adhering to established guidance for clinical trials.9
Trials and Their Design
New or better remedies are needed, particularly for patients whose disease is refractory to currently available therapies or who are dependent on cytotoxic agents or long-term glucocorticoid medication for disease control, as often seen in patients with renal involvement. These populations are not always easy to be studied in controlled clinical trials because of the heterogeneity of clinical and serologic manifestations of SLE, which may partly behave disparately. However, this is commonly an issue of study design and of reliable outcome measures (see discussion in Chapters 46 and 54), and every new drug tested in patients with SLE will have to be viewed in the context of the trial design and the population selected—with all the potential concerns that have been observed in recent clinical trials.10
Potential New Therapeutic Targets in Systemic Lupus Erythematosus
For some known and widely used drugs, such as leflunomide, methotrexate, or tacrolimus, new therapeutic opportunities may arise in SLE.11 However, in this chapter, the focus is primarily on therapeutic principles and drugs that have not yet been licensed; clinical trials in patients with SLE as listed in the U.S.-based Clinical Trials database (clinicaltrials.gov) at the time of assembling this review are summarized in eTable 56-1.
eTABLE 56-1 Summary of All Trials on Systemic Lupus Erythematosus According to the NIH-Based Registry*
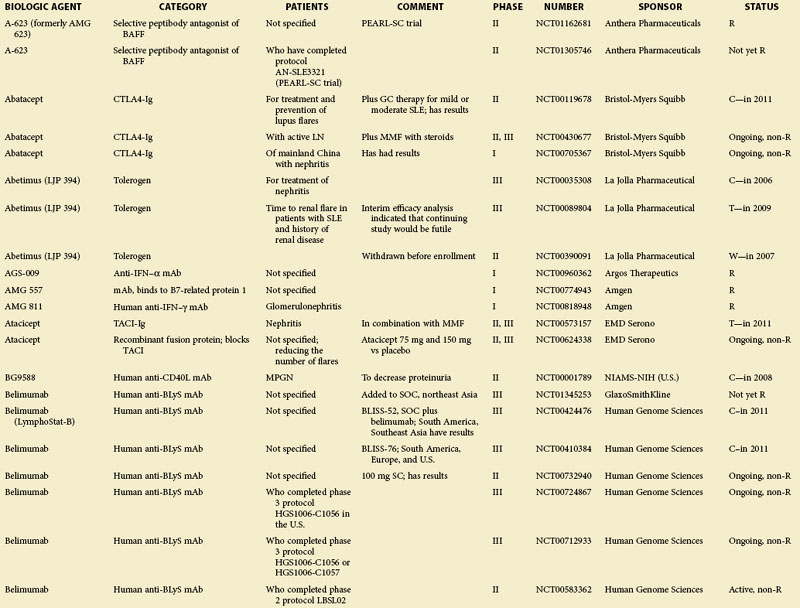
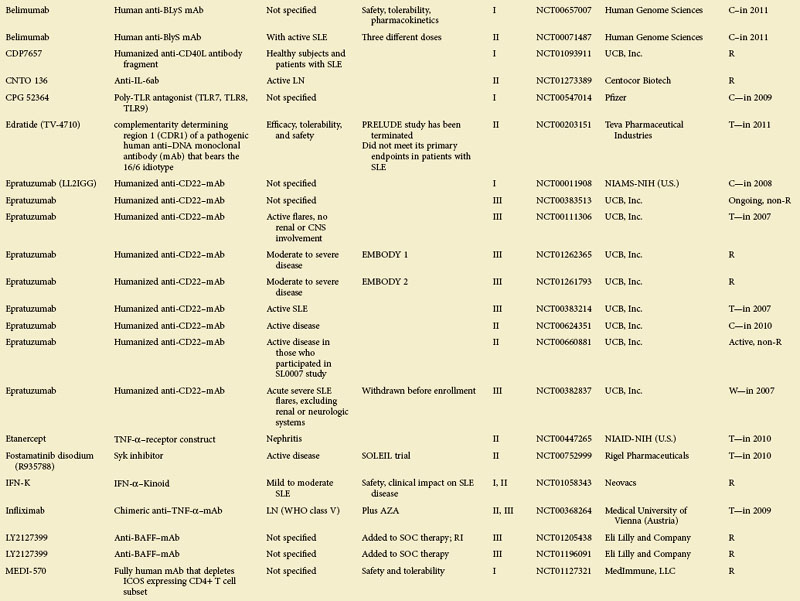
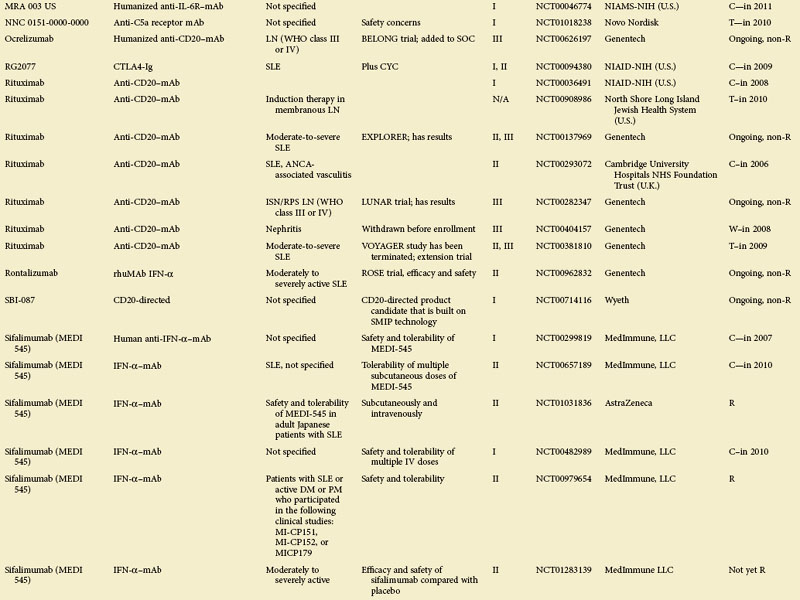
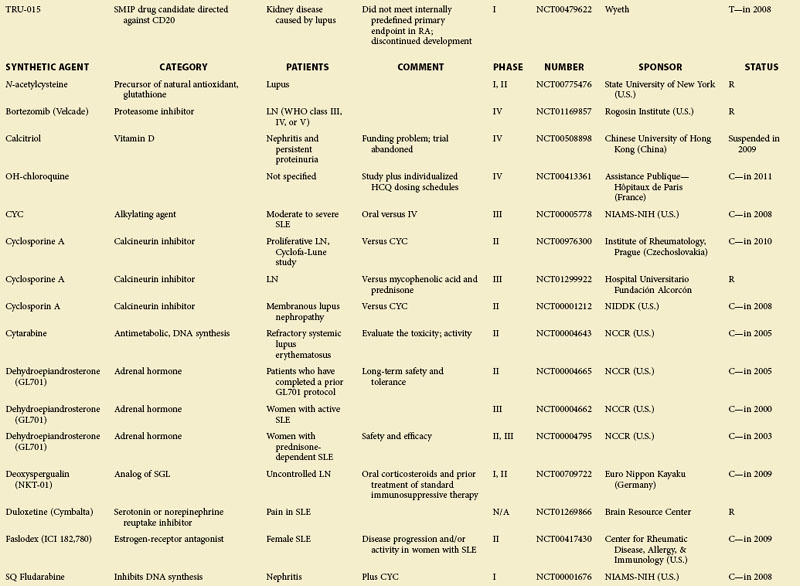
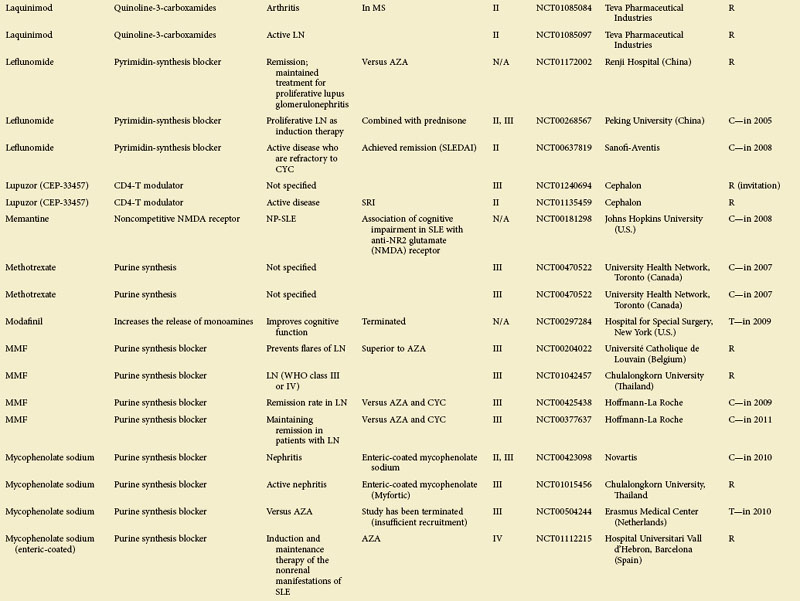
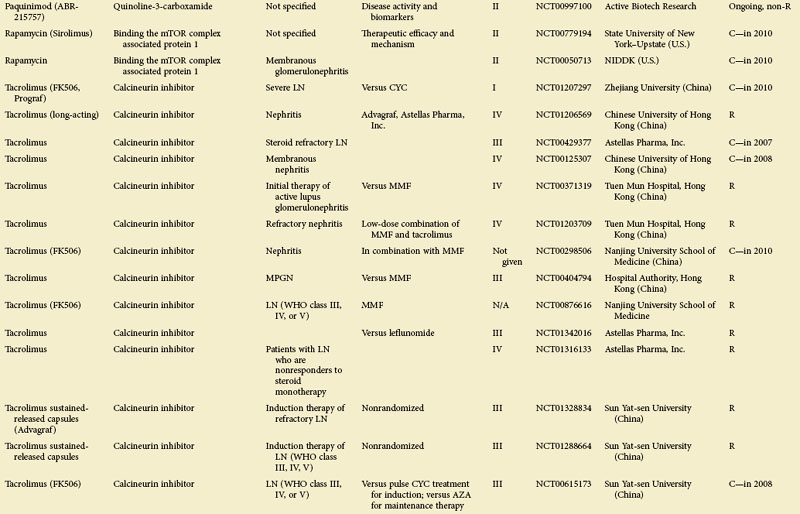
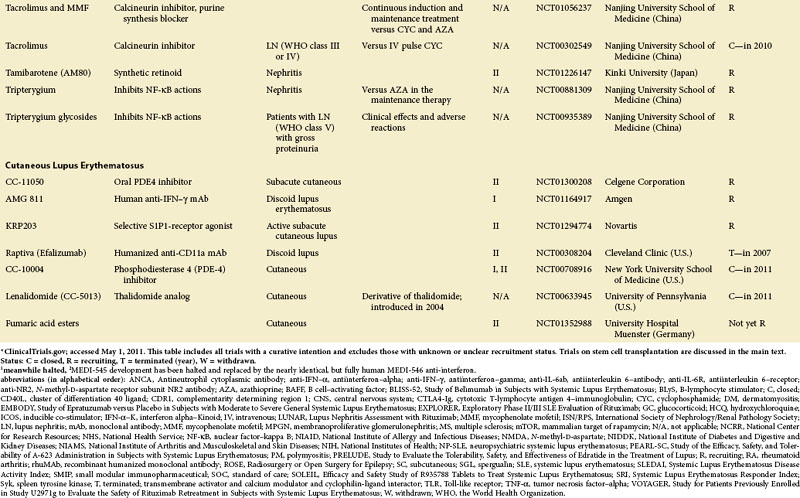
Figure 56-1 depicts major pathogenetic pathways believed to be operative in SLE and shows the close interaction of the activated innate and adaptive immune systems, as well as the subsequent inflammatory response. These pathways make up a variety of potential therapeutic targets and promising therapies that are discussed in greater detail in the following text. Table 56-2 summarizes the therapies dealt with in this chapter. The sections in this chapter and in Table 56-2 refer to targets depicted in Figure 56-1, which are numbered correspondingly.
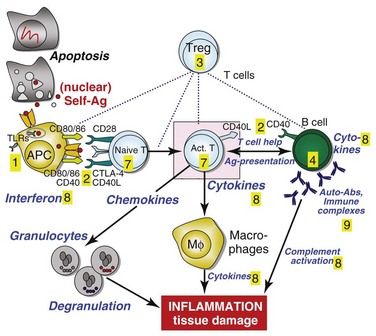
FIGURE 56-1 Overview of major pathogenetic pathways in systemic lupus erythematosus (SLE).
The activated innate and adaptive immune systems, as well as the subsequent inflammatory response, offer a variety of potential therapeutic targets and promising therapies. Current strategies aim at interfering with antigen uptake and presentation by dendritic cells (DCs) (1) and with the proper co-stimulation (2). Other attempts try to influence function and the numbers of regulatory T (Treg) cells (3) or aim to deplete or deactivate B cells (4). Stem cell transplantation (5, not shown) tries to eliminate autoreactive lymphocytes and to replace them with newly generated cells originating from undifferentiated stem cells, whereas (oral) tolerogens (6, not shown) are designed to induce tolerance in different cell types, thus abrogating autoimmune responses. Targeted therapies also aim at intracellular elements such as kinases and transcription factors (7), whereas several monoclonal antibodies target proinflammatory mediators (8). As a last resort in refractory SLE, extracorporeal procedures (9) can remove (pathogenic) autoantibodies and immune complexes. The (yellow) numbers in Figure 56-1correspond with those used in Table 56-2.
TABLE 56-2 Overview of Therapies Addressed in This Chapter
| SECTION | TITLE | SUMMARY |
|---|---|---|
| 1 | DCs and APCs | As a consequence of specifically encountering activation, exogenous (e.g., virus, bacteria) or endogenous molecules (among those apoptotic nuclear material secondary to defective clearance and potentially present in phagocytosed immune complexes), DCs are activated and/or present autoantigens. TLR7 and TLR9 are activated by RNA and DNA, respectively. Blocking these cells and receptors targets the autoimmune reaction at an early stage. |
| 2 | T cells and co-stimulation | T cells are key players in SLE; they activate autoreactive B cells and release proinflammatory cytokines. Blocking the proper co-stimulatory pathways aims at preventing T-cell activation. |
| 3 | Treg cells | Treg cells are involved in maintaining peripheral tolerance. Therapeutic strategies aim at increasing their number or functional capacity by applying specific stimuli or by applying extracorporally induced or augmented Treg cells. |
| 4 | B cells | As the producers of (pathogenic) autoantibodies, B cells are a major target of therapeutic approaches in SLE. Only a short overview is provided in this chapter. |
| 5 | Stem cell transplantation | Self-reactive lymphocytes are eliminated and replaced with newly generated cell originating from undifferentiated stem cells; key aspects of autologous and allogeneic transplantation of HSCs and MSCs are discussed. |
| 6 | Tolerogens | Inducing tolerance in DCs, T cells, or B cells might be a promising and safe approach. |
| 7 | Intracellular targets and signal transduction molecules | Most conventional immunosuppressive agents aim at intracellular mechanisms but rather nonspecifically. Targeted therapies selectively block kinases or transcription factors, or aim at messenger RNA transcripts. |
| 8 | Mediators of inflammation (interleukins, TNF-α, interferons, complement) | Antibodies against proinflammatory mediators are widely used in autoimmune diseases; however, so far, they have not shown convincing efficacy in human SLE. |
| 9 | Extracorporeal removal of autoantibodies and immune complexes | Extracorporeal removal of pathogenic autoantibodies or immune complexes is an emergency procedure in highly active SLE. |
APC, antigen-presenting cell; DC, dendritic cell; HSC, hematopoietic stem cell; MSC, mesenchymal stem cell; SLE, systemic lupus erythematosus; TNF-α, tumor necrosis factor–alpha; TLR, Toll-like receptor; Treg, T-regulator.
Antigen-Presenting Cells, Dendritic Cells, and Toll-Like Receptors
To summarize the ruling hypothesis on SLE pathogenesis: antigen-presenting cells (APCs), in general, and dendritic cells (DCs), in particular, are key players in initiating the autoimmune process. Although the eliciting antigen(s) are unknown, there has recently been a focus on autoantigens derived from apoptotic cells, since defective apoptosis mechanisms with impaired clearance of apoptotic material have been described,12 up on which nucleosomes become accessible for APCs. Although they normally have suppressive effects on DCs, they can promote cell activation when bound to the DNA-binding protein HMGB1 (high-mobility group box–1), during the late apoptotic process. As a consequence, nuclear antigens are internalized by professional APCs and, finally, may be presented to naive T cells along with the respective co-stimulatory molecules, which are also upregulated via Toll-like receptors (TLRs).13
Intracellular TLR7 and TLR9, expressed (among others) in endosomes of plasmacytoid DCs, are activated by complexes of self-protein and RNA (TLR7) or DNA (TLR9). Once activated, they promote the production of type I interferons (interferon-alpha [IFN-α]) and proinflammatory cytokines via a MyD88-dependent pathway.14 Considering the growing emphasis on the role of the interferon signature in SLE pathogenesis and since APCs and TLRs are crucial for initiating the autoimmune response by activating cells of the adaptive immune system, these cells are a tempting target for new therapies.
In contrast to cancer, in which agonists of various TLRs (and those targeting TLR7 and TLR9, in particular) are already in clinical trials,15 TLR antagonists are sought for in autoimmune disorders. Interestingly, antimalarial medications antagonize TLR9, TLR7, and TLR8, which might partly explain their effectiveness in SLE therapy. The quinazoline derivative CPG-52364 (Pfizer) specifically inhibits TLR7, TLR8, and TLR9 and inhibits SLE progression in animal models; when combined with hydroxychloroquine, it prevented anti–double stranded DNA (anti-dsDNA) antibody formation in SLE-prone mice,15 but after signals questioning its safety in healthy volunteers, phase 2 of the clinical trial was never started. The TLR7 and TLR9 antagonist IMO-3100 (Idera Pharmaceuticals) is in preclinical evaluation. TLR7 and TLR9 can also be inhibited by short DNA sequences (immunoregulatory sequences [IRS]); the compound IRS-954 (DV-1079) prevented disease progression in lupus-prone mice and reduced serum levels of nucleic acid–specific serum antibodies.15
T Cells and Co-Stimulation
Activated T cells are central players in SLE pathogenesis, since they can support B-cell activation and release cytokines that also enhance granulocyte and macrophage activity (see Figure 56-1). Preventing T-cell activation, deactivating T cells, or (at least) blocking their proinflammatory products are promising paths. Rather rude approaches with antibodies targeting all T cells (anti–cluster of differentiation 3 [anti-CD3] antibodies) have been conducted in type I diabetes and are still in use to treat renal transplant rejection16; so far, no trial has been conducted in human SLE. Interestingly, in lupus-prone mice (NZB/NZW F1 [BWF1]), nasal administration of a hamster immunoglobulin G (IgG) anti-CD3 antibody led to a reduced incidence of glomerulonephritis and decreased levels of autoantibodies, most likely by inducing a subtype of Treg cells.17 Targeting the CD4+ T-cell subtype with anti-CD4 antibodies led to amelioration of lupus in murine models (in contrast to anti-CD8 therapy, which aggravated lupus features)18 but was not pursued in humans.
Interfering with T-cell activation by blocking co-stimulation has been successfully attempted in rheumatoid arthritis (RA). When studied in SLE, abatacept (cytotoxic T-lymphocyte antigen 4–immunoglobulin [CTLA4-Ig]; Bristol-Meyers Squibb), appeared to improve musculoskeletal signs and symptoms and had a good safety profile,19 but the primary endpoint was not met, likely because of the high steroid use permitted in this study. A trial in lupus nephritis of abatacept in combination with mycophenolate mofetil (MMF) was halted,20 and another one is currently being performed by the National Institutes of Health (NIH) Immune Tolerance Network assessing abatacept in combination with cyclophosphamide ([CYC]; EuroLupus regimen) (NCT0077485). Blockade of the CD40L pathway was effective but not safe with anti–CD40 ligand antibody BG9588 (because of thrombotic complications) and safe but not effective with IDEC.21,22 CDP7657 (UCB), a pegylated Fab′ anti-CD40L compound, may be more promising, since it might not affect thrombocyte activity because of the lack of an Fc portion. In preclinical studies on mice and nonhuman primates, effective interference with the immune response was observed.23 An inducible co-stimulator (ICOS)–B7-RPI inhibitor (Amgen 557; Amgen) is currently in a phase I trial (NCT00774943) and is planned for a lupus arthritis and a subacute cutaneous lupus erythematosus (SCLE) trial; efalizumab (lymphocyte function–associated antigen [LFA] 1) was tested for cutaneous SLE but was withdrawn because of severe adverse events, namely, the occurrence of progressive multifocal leukoencephalopathy in patients with psoriasis, a disease for which SLE patients are at increased risk.24,25
Regulatory T Cells
The role of Treg cells in SLE is still under debate, but recent publications underscore that Treg cells are lower in number and have reduced suppressive capacity in active SLE.26,27 Treg cells can suppress CD4+ T-helper (Th) cells (Th1, Th2, Th17), CD8+ T cells, B cells, DCs, and a variety of other cells of the immune system. Although new Treg subtypes are continuously described,28,29 it is not entirely clear, how Treg cells exert their function and to which extent in vitro mechanisms, in fact, mimic in vivo situations or opportunities. Several different ways to suppress have been described, and more than one mechanism may be involved at one time in vivo.30 Treg cells exert suppression by releasing cytokines such as interleukin (IL)–10, they can act directly on T cells or B cells (e.g., cytolysis), or they interact with antigen presentation.31
A large body of evidence exists on beneficial effects of Treg cells in experimental models of organ-specific autoimmune diseases such as diabetes or autoimmune gastritis; interestingly, in these diseases, transforming growth factor–beta (TGF-β)–induced antigen-specific Treg cells had a much higher suppressive capacity in vivo than polyclonal (CD4+CD25+FoxP3+) naturally occurring Treg cells; the difference was greatest for suppression of Th17 cells, which are now attributed with the highest autoimmunogenic potential.32,33 Since an increased antigen specificity appeared to increase the suppressive capacity of Treg cells, interfering with antigen presentation might be a major mechanism of Treg cells in vivo.31
Since there is no known specific “lupus antigen” polyclonal Treg cells are the only available Treg population for studies in SLE. In several lupus mouse models, transfer experiments showed promising, beneficial effects; on adoptive transfer, CD4+CD25+ cells delayed disease onset (BWF1 mice), and in vitro induced polyclonal Treg cells (induction with IL-2 and TGF-β) had protective effects in lupus-like syndromes.34 In addition, nasal or subcutaneous application of autoantigens or anti-CD3 antibodies led to an increase of some Treg subtypes.35
Treg induction in humans has to be approached with caution, since murine and human Treg cells have distinct but functionally important differences. The tempting idea to boost the patients’ own Treg cells by applying exogenous triggers that proved effective in murine experiments led to a catastrophic result when a cytokine storm was elicited after the application of an anti-CD28 antibody (TGF1412 trial).36 The safer and more promising path for the future appears to be an extracorporeal induction or amplification of Treg cells; the respective techniques are already successfully tested in mice and in vitro on human cells.34
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


