Fig. 6.1
Dorsal and plantar views of clubfoot
Treatment
The two most common nonsurgical approaches in current use are the Ponseti and Montpellier (French or functional) methods, which both promote the progressive stretching of the muscles and tendons to avoid the use of the surgical soft tissue release procedure [2, 3]. Treatment by both methods ideally begins within the first few weeks after birth. The Ponseti method involves serial manipulation, casting and bracing, along with clipping of the Achilles tendon (when necessary) to obtain a corrected foot [3−5]. In contrast, the Montpellier method involves daily manipulations of the clubfoot and uses adhesive taping to maintain the correction achieved with stretching [5]. The Montpellier method requires more parental participation but allows more flexibility in the foot, whereas the Ponseti method is less time consuming but more restrictive with foot movement because of the casting and bracing. However, while these conservative treatments have improved outcomes, they do not always produce a plantar grade foot and surgery is then needed. Because of the advances with these nonsurgical treatments, untreated clubfoot is rarely seen in developed countries.
Epidemiology
Twenty to twenty-five percent of clubfoot cases are associated with a syndrome such as Distal Arthrogryposis (DA), congenital myotonic dystrophy, myelomeningocele, amniotic band sequence, Trisomy 18 and Chromosome 22q11 deletion [6−9]. The remaining 75–80 % of cases are isolated, having no other visible malformations. The birth prevalence of isolated clubfoot ranges from 1/700–1000 but varies by ethnicity from a high of 6.8/1000 in Hawaii to a low of 0.87/1000 in Japan (Table 6.1) [10−24]. Bilateral involvement is found in half of all cases, while in the unilateral cases, the right foot is affected more often than the left [22, 25]. Interestingly, males are affected twice as frequently as females [25, 26]. Clubfoot is a complex birth defect in which both genetic and environmental factors, presumably in combination as well as separately, contribute to the etiology [27].
Table 6.1
Birth rates of clubfoot
Population | Incidence/1000 live births |
|---|---|
Australia: Aboriginal [22] | 3.49 |
Australia: Cauasians [22] | 1.11 |
Belgium [21] | 1.6 |
Denmark [20] | 1.2 |
Hawaii [19] | 6.8 |
India [18] | 0.9 |
Japan [17] | 0.87 |
Malawi [16] | 2 |
Papua New Guinea [15] | 2.7 |
Sweden [14] | 1.4 |
Uganda [13] | 1.2 |
US [12] | 1.29 |
The Role of Environmental Factors in Clubfoot
Many environmental factors have been suggested to contribute to clubfoot but few have been consistently identified. Seasonal variation has been observed in some studies with higher birth prevalence in winter (December–March), while other studies have found no correlation [12, 28−31]. Similarly, sulfonamides and abortifactent agents have been inconsistently identified [32, 33]. While maternal folic acid supplementation decreases the birth prevalence of neural tube defects, only a small reduction in isolated clubfoot has been found on a population basis [24, 34]. There is little evidence to suggest that folic acid deficiency contributes to clubfoot.
Maternal smoking is the only environmental factor consistently associated with clubfoot [10, 35−39]. Mothers who smoke during pregnancy have an increased risk of having a child with clubfoot and this risk increases in a dose-dependent pattern (Relative Risk: 1.3–2.2) [37−39]. Maternal smoking during pregnancy and a positive family history of clubfoot increased the risk of clubfoot [10, 38].
Genetic Epidemiology of Clubfoot
Evidence for a genetic etiology for clubfoot comes from studies showing (1) aggregation of clubfoot in families, (2) increased risk with number of affected siblings, (3) heritability for clubfoot of 72 %, and (4) higher concordance in monozygotic than dizygotic twins (32.5 vs. 2.9 %) [15, 40−42]. Segregation analyses from different populations support a multifactorial/oligogenic model in which variation in multiple genes influenced by environmental factors play a causal role [17, 24, 41, 43−46]. It is now well accepted that genetic variation contributes to clubfoot, however the mechanism(s) remain to be defined. The generally accepted model is that of multifactorial inheritance with a threshold effect [47].
Genes and Environment: Tobacco Metabolism and Clubfoot
Cigarette smoke consists of more than 4000 chemical compounds that are metabolized through the xenobiotic metabolism [48]. The identification of maternal smoking as a risk factor suggests that variants in genes involved in xenobiotic metabolism may contribute to clubfoot. Biotransformation through this pathway can detoxify a compound or it can create a more toxic/reactive intermediate metabolite that can create adducts. Adducts can bind DNA or proteins altering transcription and replication that could affect normal development [49]. N-acetyltransferase (NAT) genes are obvious candidate genes because they acetylate toxins including free radicals found in cigarette smoke. Variants in NAT1 and NAT2 were genotyped in a clubfoot dataset composed of both multiplex families (family history of clubfoot) and simplex (no family history) trios [50]. More slow acetylators were found in cases suggesting a potential link between clubfoot risk and slow acetylation [50].
Given the importance of this pathway, eight xenobiotic metabolizing genes, including NAT1 and NAT2 were interrogated in a subsequent study [51]. rs1048943/CYP1A1 was significantly associated with nonHispanic white (NHW) multiplex families (p = 0.009) [51]. It was hypothesized that the presence of variation in multiple genes in this pathway would increase the risk over variation in a single gene. Indeed, the allelic combination comprised of rs105740/EPHX1 and rs1799929/NAT2 was found to be associated with clubfoot (p = 0.007). In addition, evidence for maternal and fetal genotypic effects were found for two SNPs in CYP1A2. A significant maternal genotypic effect was found for rs11854147/CYP1A2 with a relative risk of 1.24 (95 % CI: 1.04–1.44, p = 0.03) and a significant fetal genotypic effect for rs2470890/CYP1A2 with a relative risk of 1.33 (95 % CI: 1.13–1.54, p = 0.01) [51]. These findings suggest that xenobiotic metabolism genes are unlikely to play a major role in clubfoot; however, perturbation of this pathway may play a contributory role.
Other Approaches to Gene Identification in Clubfoot
Given that there is a genetic component to clubfoot, studies have focused on identifying the clubfoot genes mainly using genome scans and candidate gene approaches. In brief, genome-wide scans utilize polymorphic markers such as SNPs and copy number variants (CNVs) spanning the genome to identify linked and/or associated variants/regions with the phenotype of interest. In contrast, the candidate gene approach uses prior knowledge to identify genes for consideration. In addition to genes in the xenobiotic pathway, genetic investigations of clubfoot have included analysis of genes that (1) play a biological role in hindlimb development, (2) cause a clubfoot phenotype when altered in an animal model and/or (3) are deleted, duplicated or mutated in a syndrome that has multiple malformations including clubfoot (Fig. 6.2). The following sections discuss the potential clubfoot genes and variants that have been identified.
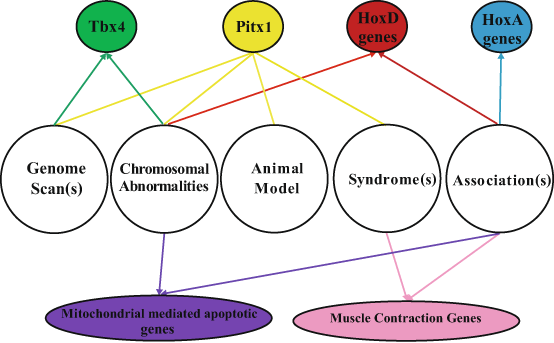
Fig. 6.2
Methods used to identify clubfoot genes and identified genes
Hindlimb Development
Hindlimb morphogenesis is a multifaceted process involving cell proliferation, migration, patterning and programmed cell death, all of which are regulated by numerous factors such as growth and transcription factors and signaling molecules [52−57]. Hindlimb development begins between the fourth and fifth week of gestation with swelling along the lateral plate mesoderm. By the middle of the fifth week, the limb bud becomes pronounced and it is the regulation of three major axes: proximal-distal (PD, from knee to digits), anterior-posterior (AP, from the big toe to the little toe) and dorsal-ventral (DV, from the top of the foot to the bottom of the foot) that transforms the limb bud into a mature limb. Chondrification begins by the end of the sixth week; osteogenesis begins in the seventh week and is present in all the long bones by the twelfth week. Myoblasts aggregate and form large muscle masses in each limb bud differentiating into dorsal and ventral components as the long bone forms. As development progresses through week 6 and 7, programmed cell death occurs removing the interdigital webbing and freeing the individual toes [52]. During the final stages of limb development in weeks 8–11, the foot begins to rotate from an inward apposing position to a plantar position [58]. This complex process is regulated by a large number of genes which when perturbed could contribute to a clubfoot.
PITX1
Evidence for a role of PITX1 , paired-like homeodomain 1, has recently been reported [59]. PITX1, a bicoid-related homeodomain transcription factor, was first identified in a genome-wide linkage study (5q31: LOD: 3.31) in a five-generation clubfoot family with nine affected individuals [59]. A single missense mutation (E310K) in PITX1 segregated with the clubfoot and was not present in 500 controls [59]. The mutation is located in a highly conserved homeodomain and reduces wild-type PITX1 activity in a dose-dependent manner suggesting a dominant-negative effect on transcription [59]. Although this was the first evidence for a hindlimb-specific gene variant in clubfoot, members of this family had other lower-limb malformations including patellar hypoplasia, oblique talus, preaxial polydactyly suggesting that the mutation causes a syndromic form of clubfoot. This is further supported by the identification of PITX1 deletions in three other individuals with isolated lower-limb malformations including polydactyly [60].
Another study provides additional evidence for a role of PITX1 in isolated clubfoot. A copy number analysis of 40 familial clubfoot probands identified a 241 kb microdeletion involving PITX1 in one individual [61]. The authors suggested PITX1 haploinsufficiency as a cause for isolated clubfoot and cited support based on decreased PITX1 activity from the E310K/PITX1 mutation [59, 62]. Additional supporting evidence for a role of PITX1 haploinsufficiency comes from the Pitx1 haploinsufficient mouse; approximately 8.9 % of these mice have a clubfoot-like phenotype [61]. This is the first animal model to closely mimic human clubfoot. However, the Pitx1 haploinsufficient mouse does differ from the human clubfoot in a number of ways: (1) females are affected more often than males, the opposite of the human condition, (2) there is peroneal artery hypoplasia, and (3) the tibial and fibular bone volumes are reduced. Nevertheless, the role of PITX1 in isolated clubfoot remains intriguing. Other families may provide more etiologic and phenotypic information.
TBX4
T-box transcription factor 4, TBX4 , a direct transcriptional target of PITX1 has been implicated in clubfoot [63]. Like PITX1, TBX4 is also specifically expressed in the hindlimb and involved in limb muscle and tendon patterning [64, 65]. One study screened 66 familial clubfoot probands for genomic copy-number variants and found a 17q23.1q23.2 microduplication in three probands. The microduplication segregated in the families but showed reduced penetrance. Individuals with the microduplication also had mild short stature, developmental hip dysplasia and subtle skeletal abnormalities such as short wide feet. While the authors suggest that the 17q23.1q23.2 microduplication is a common cause for familial isolated clubfoot, only a few families were identified. A second study identified only one multiplex family out of 605 clubfoot families with a small 350 kb 17q23.1q23.2 microduplication [66]. This microduplication segregated with the clubfeet and the short, wide feet, once again supporting variable expressivity with this microduplication [66]. Therefore, the role of TBX4 in the etiology of isolated clubfoot remains to be elucidated but appears to cause a syndromic form of clubfoot. Even more intriguing is the possible interactive role of genetic variants in the TBX4-PITX1 pathway, which should be thoroughly interrogated .
Muscle Contraction Genes
Clubfoot can occur as part of a number of Mendelian syndromes, which suggests that variation in the genes underlying these disorders could have an etiologic role in the isolated form. This is the paradigm that led to the identification of genetic variation in IRF6 and nonsyndromic cleft lip and palate [67]. The Distal Arthrogryposis (DA) syndromes, for example, are characterized by congenital joint contractures and many include clubfoot [68]. Currently, there are nine different types of DA and clubfoot occurs in four. Mutations in muscle contraction genes, MYH3, TNNT3, TNNI2, TPM2, MYBPC1 and TPM8, cause DA1, DA2A, DA2B and DA7 [68]. One study sequenced the exons of MYH3, TNNT3 and TPM2 in 20 clubfoot probands from multiplex clubfoot families [69]. While rare exonic variants were identified, none segregated in all affecteds in each family. The authors suggested that these genes do not contribute to clubfoot, however the regulatory regions were not sequenced [69]. Similarly, sequencing of the exons, splice sites and predicted promoters of embryonic and perinatal myosin genes, MYH2, 3, 7 and 8 did not find any associations [70].
A more recent study interrogated fifteen muscle contraction genes in 224 families with a family history of clubfoot and 357 families with no family history and included upstream and downstream genomic regions in order to capture variation in the regulatory regions [71]. Several positive associations were found in the single SNP, haplotype and gene-gene interaction analyses for both the nonHispanic white (NHW) and Hispanic groups. Interestingly, the most significant associations involved SNPs located in potential regulatory regions, in TPM1 and TPM2 [71]. A follow-up study addressed the functional implications of these variants [72]. The TPM2 SNPs, rs2025126 and rs2145925, altered nuclear protein binding and changed promoter activity in C2C12 mouse muscle cells. Interestingly, while the rs4075583/TPM1 alternative allele showed an alteration in nuclear protein binding it did not affect promoter activity. However, when rs4075583 was analyzed in the context of a ~ 1.7 kb eight SNP haplotype, varying degrees of promoter activity were observed depending on allelic combination. Promoter activity was decreased when rs4075583 alternate allele was present in the haplotype [72]. These results suggest the importance of transcriptional regulation in muscle contraction genes as a mechanism for clubfoot .
Homeobox A and D Genes
In order to identify chromosomal regions/genes that contribute to human malformations, Brewer et al. combined clinical reports to identify chromosomal abnormalities, deletions and duplications, in syndromic clubfoot [8, 9]. For clubfoot, this included six large chromosomal deletion regions (2q31-33, 3q23-24, 4p16-14, 7p22, 13q33-34 and 18q22-23) and two duplication regions (6q21-27 and 10p15-11) [8, 9]. Interrogation of the 2q31-33 deletion region identified associations with variations in CASP8, CASP10 and CFLAR [73, 74]. These genes are involved in the mitochondrial-mediated apoptotic pathway which is consistent with the key role that apoptosis plays in limb and muscle development. In a follow-up study, interrogation of five additional mitochondrial-mediated apoptotic genes (CASP9, CASP3, APAF1, BCL2 and BID) identified suggestive associations between variation in each gene and clubfoot [75].
Also present in the 2q31-33 deletion region is the Homeobox gene cluster D (HOXD). Genes in the HOXD cluster direct limb and muscle patterning during development. This cluster is functionally redundant with the Homeobox A gene cluster (HOXA) located on chromosome 7p15; this region was not identified in the report of Brewer et al. [76−78]. Mutations in both HOXA and HOXD have been associated with syndromes that involve limb abnormalities but not clubfoot [79, 80]. The HOXA genes are also known to regulate the synchronized development of muscles, tendons and cartilages [78]. Interrogation of these genes in the clubfoot nonHispanic and Hispanic multiplex and simplex families identified associations with SNPs in both gene clusters [74]. One SNP, rs3801776, located in the basal promoter of HOXA9, gave the strongest association with clubfoot in both discovery and validation clubfoot groups (p = 0.004 and p = 0.03, respectively) [74]. The ancestral allele of rs3801776 created a nuclear protein binding site that increased promoter activity [72]. These results suggest that perturbation in genes involved in limb and muscle patterning and development play a role in clubfoot. Again, the association was with regulatory SNPs suggesting that gene expression plays an important role in clubfoot.
Summary
Clubfoot is a well-recognized birth anomaly for which etiologic insights are only now being identified. Genome-wide linkage studies are only successful if there are sufficient numbers of large families to overcome any heterogeneity issues. While multiplex clubfoot families do exist, and it has been suggested that clubfoot fits a single major gene model [46, 81, 82], most families have only 2 or 3 affected individuals. Moreover, extensive heterogeneity would be expected given the complexity of hindlimb development. As a result, most linkage studies of clubfoot have not been successful [81, 83]. Evidence for involvement of PITX1 and TBX4 , both hindlimb-specific genes, have been found in only a few clubfoot families/cases and not confirmed in association studies, suggesting that variation in these genes is not involved in the majority of cases of clubfoot [27, 66]. Genome-wide associations studies are most successful when the disease in question is due to common variation and the dataset is sufficiently large. Lack of a large dataset can be circumvented to some extent by focusing on candidate genes. This approach has led to the identification of associations between clubfoot and variation in several genes, including HOXA9 and the muscle contraction genes TPM1 and TPM2. While potential mechanisms have been described for clubfoot for these genes, independent verification is still needed. Given the paucity of large families or the lack of large datasets, clubfoot genetic studies should benefit from next-generation approaches, such as whole exome and genome sequencing. Families can be examined individually and in concert with other families to identify genes and pathways, which appear to underlie the development of clubfoot. Results of these studies will then require verification in animal models and functional studies. The challenge is great but the payoff is also large as it will provide insight into biological mechanisms causing clubfoot, and in some cases families may benefit from personalized recurrence risk information.
Related posts:
 The Genetic Architecture of Idiopathic Scoliosis
Molecular Genetics of Congenital Multiple Large Joint Dislocation
Overview of Next Generation, High-Throughput Molecular Genetic Methods
Neurofibromin in Skeletal Development
DMP-1 in Postnatal Bone Development
Genetic and Environmental Interaction in Malformation of the Vertebral Column
The Genetic Architecture of Idiopathic Scoliosis
Molecular Genetics of Congenital Multiple Large Joint Dislocation
Overview of Next Generation, High-Throughput Molecular Genetic Methods
Neurofibromin in Skeletal Development
DMP-1 in Postnatal Bone Development
Genetic and Environmental Interaction in Malformation of the Vertebral Column
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


