Chapter 73 Hemophilia and Pigmented Villonodular Synovitis
Hemophilia
The clinical severity of the hemophiliac patient is inversely proportional to the level of plasma coagulant factor activity. When factor VIII:C or factor IX activity levels are less than 1% of normal (severe hemophilia), frequent spontaneous bleeding (no overt trauma) into joints and soft tissues is expected, and prolonged, profuse bleeding typically accompanies trauma or surgery. Factor VIII:C or factor IX activities ranging from 1% to 5% of normal levels signify a more moderate clinical course, characterized by occasional spontaneous bleeding and excessive bleeding with surgery or trauma. Factor VIII:C and factor IX levels greater than 5% of normal do not predispose patients to spontaneous bleeding events, although excessive bleeding with surgery or trauma can occur. Severe (<1% factor VIII:C activity) disease has been noted to occur in about 60% of patients with hemophilia A, whereas severe disease has been noted in about 20% to 45% of patients with hemophilia B (Table 73-1).
Table 73-1 Hemophilia Clinical Severity
| Class | Factor VII and IX Levels,% Normal | Bleeding |
|---|---|---|
| Mild | >5 | Bleeding only with significant trauma or surgery |
| Moderate | 1-5 | Occasional spontaneous bleeding; usually bleeding only with mild trauma |
| Severe | <1 | Frequent spontaneous bleeding |
Laboratory Evaluation
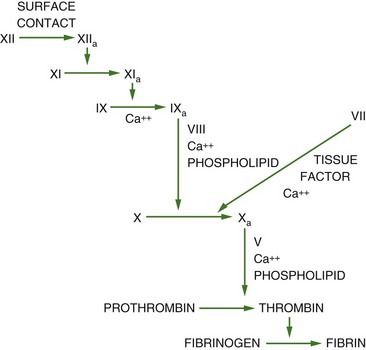
Normal factor VIII:C and factor IX activities range from 50% to 150%. A significantly prolonged activated partial thromboplastin time is the most common abnormal coagulation laboratory assay in hemophilia A and hemophilia B because factor VIII:C and factor IX lie within the intrinsic pathway of coagulation (Fig. 73-1). In North America, the one-stage clotting assay is performed on patient plasma to determine the exact levels of clotting factor VIII:C and factor IX activity. This assay is based on the activated partial thromboplastin time and is reproducible, technically simple and automated, and inexpensive.
Factor Replacement Therapy
Commercially available clotting factor concentrates licensed in the United States are listed in Table 73-2. No difference in efficacy has been noted among these concentrates, but there may be perceived and real potential safety advantages of recombinant products over plasma-derived products, so more than 90% of replacement therapy for hemophilia A and hemophilia B in the United States is accomplished with recombinant products. With the advent of virtually safe viral attenuated clotting factor replacement therapies, fresh-frozen plasma and cryoprecipitate are used rarely in the treatment of hemophilia A and hemophilia B. Rather, fresh-frozen plasma is the preferred replacement therapy for the rare clotting factor deficiencies for which no specific replacement product exists (e.g., factor XI deficiency). Cryoprecipitate is hardly used in the United States as a replacement product except in patients with afibrinogenemia.
Table 73-2 Factor VIII and Factor IX Concentrates Available in the United States
| Virucidal Method | Type/Name of Product | Manufacturer |
|---|---|---|
| Factor VIII | ||
| Immunoaffinity chromatography | Advate | Baxter-Immuno |
| Refacto | Wyeth-Ayerst | |
| Kogenate-FS | Bayer | |
| Helixate-FS | Bayer* | |
| Immunoaffinity chromatography and pasteurization (60° C, 10 hours) | Monoclate P | ZLB-Behring |
| Immunoaffinity chromatography/solvent detergent, heat treatment (25° C, >10 hours) | Hemophil M | Baxter-Immuno |
| Monarc M | Baxter-Immuno† | |
| Affinity chromatography, solvent detergent, and terminal heating (80° C, 72 hours) | Alphanate SD | Griffols |
| Solvent detergent (TNBP/polysorbate 80) | Koate-HP | Bayer |
| Factor IX | ||
| Affinity chromatography and ultrafiltration | BeneFIX | Wyeth-Ayerst |
| Dual-affinity chromatography, solvent detergent (TNBP/Polysorbate 80), and nanofiltration | AlphaNine SD | Griffols |
| Immunoaffinity chromatography, solvent detergent (sodium thiocyanate), and ultrafiltration | Mononine | ZLB-Behring |
| Dry heat (80° C, 72 hours) | Konyne 80 | Bayer |
| Dry heat (68° C, 144 hours) | Proplex T | Baxter-Immuno |
† Prepared from volunteer donor plasma provided by the American Red Cross.
Pathophysiology of Hemophilic Arthropathy
Hemophilia results in spontaneous bleeding into the knee. Repeated bleeds first cause a synovitis, which leads to articular cartilage destruction. The mechanism of joint damage is thought to be multifactorial. Chemical and inflammatory factors arise from the occurrence of bleeds in the joint. These two factors are followed by physical and biomechanical degradation of the articular cartilage.28
When bleeding has occurred in the knee, hemosiderin-stained deposits appear in the synovium (Fig. 73-2). Evidence indicates that iron deposits in the synovium stimulate the production of cytokines. These proteins damage the articular cartilage. It seems likely that phagocytosis by synovial cells leads to stimulation of cytokine production. Roosendal and coworkers56 showed significantly higher production of interleukin-1, interleukin-6, and tumor necrosis factor in cultures of hemophilic synovial tissue compared with normal tissue. In addition, supernatant fluid from these cultures showed greater catabolic activity. Proteoglycan synthesis has been shown to be inhibited by these supernatants. Arnold and Hilgartner1 have shown high levels of acid phosphatase and cathepsin D in hemophilic synovial tissue samples. Stein and Duthie62 studied the histochemistry of synovium retrieved from hemophiliac individuals undergoing reconstructive surgery. They showed hemosiderin deposits in the synovium and articular cartilage. They postulated that the hypertrophic synovial membrane led to the release of lysosomal enzymes, such as cathepsin D, resulting in articular cartilage breakdown.
Radiology of Hemophilic Arthropathy of the Knee
Radiographic evaluation is the primary diagnostic tool used to evaluate the knee in patients with hemophilia. Standing anteroposterior and lateral views should be taken. In addition, skyline or Merchant views add valuable information about the presence of patellofemoral disease. Magnetic resonance imaging (MRI) has been shown to be valuable in the evaluation of knees with minimal x-ray changes.4,75 Often MRI shows evidence of synovial hypertrophy and other pathology before the standard x-ray. MRI may have particular value in the treatment of children with early disease who are candidates for synovectomy.43 Wilson and colleagues74 proposed the use of diagnostic ultrasound as an adjunct in the decision-making process for soft tissue swelling around the hemophilic joint. Ultrasound may aid in the diagnosis of pseudotumors.
Two radiographic classification systems are in use today. Arnold and Hilgartner1 described a five-stage system in 1977. This logical, simple system easily divides the presentation of joint changes into stages that have surgical significance. In 1979, Pettersson and associates46 described an eight-stage system for grading arthropathy. Although this system is more detailed, it is not as commonly used today as the five-stage system of Arnold and Hilgartner (Table 73-3):
Table 73-3 Arnold and Hilgartner’s Radiographic Classification of Hemophilic Arthropathy
| Stage | Radiographic Finding |
|---|---|
| 0 | Normal knee |
| I | Soft tissue swelling |
| II | Soft tissue swelling, osteopenia, epiphyseal overgrowth, no narrowing of joint space |
| III | No significant narrowing of joint space, subchondral cysts, osteopenia |
| IV | Destruction of cartilage and narrowing of joint space |
| V | End stage, with destruction of joint and gross bony changes |
Data from Arnold WD, Hilgartner MW: Hemophilic arthropathy. J Bone Joint Surg Am 59:287, 1977.
The classification system of Pettersson and associates46 consists of eight different radiographic signs: osteoporosis, epiphyseal enlargement, joint margin erosion, irregular subchondral surface, joint incongruity, subchondral cysts, joint space narrowing, and angular deformity. Each sign is graded 0 points (no change), 1 point (slight change), or 2 points (severe changes). The total score yields a particular stage. Greene and colleagues24 compared both of these systems and found that the Pettersson system was more accurate. A new system of four signs and seven points was proposed. This system was shown to be more accurate; however, the Arnold and Hilgartner system1 remains the most commonly used system in the United States today.
Related posts:
 Cemented Total Knee Arthroplasty: The Gold Standard
Cemented Total Knee Arthroplasty: The Gold Standard
 Management of Extra-articular Deformity in Total Knee Arthroplasty With Navigation
Management of Extra-articular Deformity in Total Knee Arthroplasty With Navigation
 Computer-Assisted Navigation: Minimally Invasive Surgery for Total Knee Replacement
Computer-Assisted Navigation: Minimally Invasive Surgery for Total Knee Replacement
 Distal Realignment of the Patellofemoral Joint: Indications, Effects, Results, and Recommendations
Distal Realignment of the Patellofemoral Joint: Indications, Effects, Results, and Recommendations
 Anterior Cruciate Ligament Reconstruction With Central Quadriceps Free Tendon Graft
Anterior Cruciate Ligament Reconstruction With Central Quadriceps Free Tendon Graft
 Osteochondritis Dissecans
Osteochondritis Dissecans
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


