Chapter 34 Hematologic and Lymphoid Abnormalities in SLE
Anemia
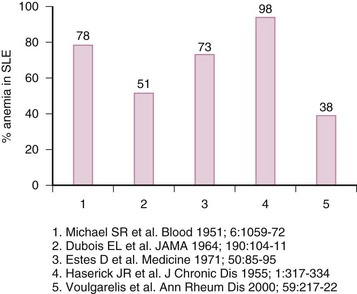
Most patients with SLE will develop anemia at some point throughout the course of the disease (eFig 34-1). The most prevalent type is anemia of chronic disease (ACD); however, iron deficiency anemia (IDA), autoimmune hemolytic anemia (AIHA), drug-induced myelotoxicity, and anemia of chronic renal failure are not uncommon. Box 34-1 illustrates the types of anemias in SLE by decreasing order of frequency.
Box 34-1
Classification of Anemia in Systemic Lupus Erythematosus
Causes of Anemia in Systemic Lupus Erythematosus (SLE)
Anemia of Chronic Disease
A large, single-center prospective study of 132 anemic patients with SLE identified ACD as the most prevalent variant, accounting for 37% of all patients.1 Although generally mild (mean hemoglobin [Hgb] 9.9 ± 1.3 g/dL), 50% of patients with Hgb less than 8 g/dL from the entire cohort fell in the category of ACD. The same study reported that patients with ACD had higher disease activity as measured by European Consensus Lupus Activity Measurement (ECLAM) scores, compared to those with other types of anemias (P = 0.01). This study speculated that higher ECLAM scores might reflect the higher prevalence of lupus nephritis in the ACD group (57%). This type of anemia is likely to persist for prolonged periods in contrast to others; over 50% of patients may still be anemic after 3 years of follow-up.1
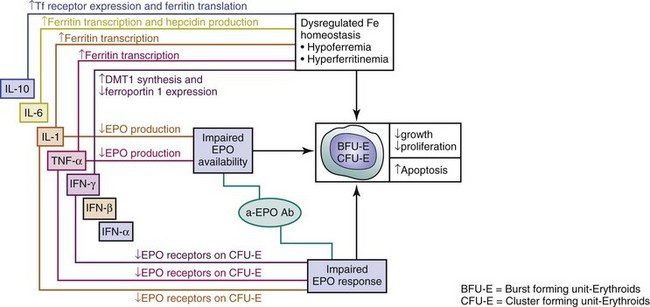
ACD in SLE represents a hypoproliferative state with several dimensions (Figure 34-1):
(a) Abnormal iron metabolism with sequestration in macrophages, leading to hypoferremia and unavailability to erythropoietic progenitors for Hgb synthesis
(b) Decreased erythropoietin (EPO) supply to red cell progenitors
(c) Increased resistance of responder cells to the proliferative actions of EPO
Iron metabolism was investigated in 11 patients with SLE using radioactive iron (59Fe).2 Iron use was decreased in 7 patients, with increased uptake over the spleen and liver where it was stored, instead of use for Hgb synthesis. Plasma iron turnover, on the other hand, was elevated in most patients. Additionally, the life span of erythrocytes was reduced in the absence of hemolysis. Iron metabolism in ACD at large is currently accepted as mainly altered by an overproduction of the acute phase protein, hepcidin.3 This cysteine-rich cationic peptide is the main negative regulator of intestinal iron absorption, transport across the placenta, and iron release from macrophages. Its production is mainly driven by interleukin-6 (IL-6). Although IL-6 levels are reportedly higher in anemic compared to nonanemic SLE subjects, and an inverse correlation between IL-6 and Hgb levels was shown,4 the contribution of the hepcidin pathway in SLE-associated ACD has not been explored. Prohepcidin levels have been evaluated and shown to have no association with Hgb levels in anemic patients with SLE.5 However, prohepcidin levels, it is argued, may not associate with serum iron status and may not accurately reflect mature circulating hepcidin.5
Insufficient EPO supply to hematopoietic progenitors and enhanced resistance of those cells to the proliferative effects of EPO are the main culprits in SLE-associated ACD (see Figure 34-1). A decreased EPO supply may reflect lower production or enhanced turnover. Impaired production of EPO has been reported in patients with SLE-associated ACD; 42% showed at least 25% lower EPO levels, compared with controls for levels of Hgb, and the slope of EPO response to anemia was significantly blunted (P = 0.01).1 Lower EPO production may be the result of the inhibitory action of cytokines such as interleukin-1 alpha (IL-1α), IL-6, tumor necrosis factor-alpha (TNF-α), interferon alpha (IFN-α), interferon beta (IFN-β), interferon gamma (IFN-γ), and transforming growth factor–beta (TGF-β), all of which are largely implicated in SLE. Specifically, Faquin and colleagues6 reported that IL-1, TNF-α, and TGF-β inhibited EPO production from the hepatoma line of Hep3B cells at the level of EPO messenger ribonucleic acid (mRNA). Jelkmann and others7 additionally showed that IL-1β inhibited EPO production in isolated serum-free perfused rat kidneys. In patients with lupus nephritis, effector cluster of differentiation 4 (CD4+) T lymphocytes and macrophages infiltrate the renal interstitium and produce cytokines with inhibitory effects on EPO production.8 Since such patients frequently display ACD, this mechanism can certainly explain suppressed EPO levels in this subset.
The presence of antibodies against EPO (anti-EPO antibodies) constitutes an alternative explanation for impaired EPO supply; they may bind and neutralize EPO before binding its receptors on target hematopoietic cells. The prevalence of anti-EPO antibodies in unselected patients with SLE is 15%; this is significantly higher in patients with Hgb less than 10 g/dL, compared with nonanemic patients (29% versus 9%; P < 0.05), and in those with ACD-related SLE (38%).1 Additionally, anti-EPO antibody titers are higher in patients with severe anemia, compared with those with moderate anemia, and patients with such antibodies have higher disease activity compared to those without them.1 The presence of ACD and the severity of ECLAM scores independently predicted the prevalence of anti-EPO antibody (odds ratio [OR] = 3.1, P = 0.04 for the presence of ACD, and 1.27 per each ECLAM point, P = 0.055).1 Despite these observations indirectly supporting a neutralizing effect of these antibodies on EPO, it remains unclear whether antibodies to endogenous EPO have a direct pathogenic role in the induction of ACD in SLE. No correlation between anti-EPO antibodies and EPO levels has been demonstrated; however, the possibility of interference of such antibodies in the measurement of EPO cannot be excluded as a major confounder. Anti-EPO antibodies binding to EPO in the form of immunoglobulin G (IgG) complexes may stabilize and prolong EPO half-life, which explains its accumulation, despite the fact that it may not be biologically active.9 A rare scenario, in which an inverse association between antibodies to endogenous EPO, EPO levels, and Hgb has been reported, is pure red cell aplasia (PRCA) in SLE. In these patients, immunosuppression decreases antibody titers and corrects Hgb levels.9 Another similar scenario is the induction of secondary PRCA in patients with end-stage renal disease who receive exogenous EPO; temporary withdrawal and immunosuppression successfully decrease antibody levels and treat the anemia. Despite the ability of SLE-associated ACD to respond well to EPO, little rationale exists for such use given the reasons previously stated. Additionally, prior in vitro studies on T and B cells in dialysis patients receiving recombinant human erythropoietin (rHuEPO) suggested that it might augment immune responses.8
Enhanced resistance of red cell precursors to the proliferative effects of EPO has also been incriminated in the pathogenesis of SLE-associated ACD (see Figure 34-1). Downregulation of surface EPO receptors and the induction of apoptosis in erythroid precursors have been demonstrated by IL-1, TNF-α, and IFN-γ. Proinflammatory cytokines and chemokines exert direct effects on erythropoiesis beyond their involvement in EPO elaboration, amplifying the risk of ACD (see Figure 34-1). TNF-α acts directly on the less mature burst-forming units–erythroid (BFU-E), whereas its effect on colony-forming units–erythroid (CFU-E) is indirect, via the induction of IFN-β release from bone marrow (BM) stromal cells.10 IL-1 inhibits colony formation of CFU-E indirectly by upregulating the production of IFN-γ by T cells.10 IFN-α inhibits BFU-E directly and CFU-E indirectly through accessory cells.10
Iron Deficiency Anemia
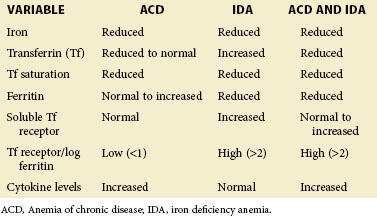
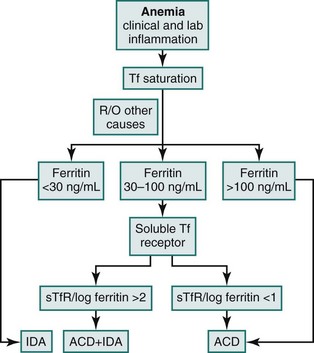
Laboratory parameters and an algorithm differentiating ACD, IDA, or a true coexistence of IDA with ACD are provided in eTable 34-1 and eFig 34-2, respectively.
eTABLE 34-1 Laboratory Parameters Differentiating Anemia of Chronic Disease from Iron Deficiency Anemia in Systemic Lupus Erythematosus
Immune-Mediated Hemolytic Anemias
Autoimmune Hemolytic Anemia
Antibody-mediated erythrocyte damage by complement-dependent or complement-independent mechanisms is the third most common cause of anemia in SLE; it is reported in 5% to 14% of patients with this disease.11 Approximately two thirds of patients with SLE-associated AIHA exhibit symptoms at the onset of SLE,12 and 41% to 90% may already be taking immunosuppressive medications at the time of diagnosis. AIHA corresponds with large acute decreases in Hgb levels. The severity of anemia is highest in AIHA, compared with other types in SLE; in a large prospective study, mean Hgb with AIHA was 8.99 ± 1.5 g/dL, compared with 10.9 ± 0.9 for IDA, 9.94 ± 1.3 for ACD, and 9.64 ± 1.8 for the group of other causes (P < 0.001).1 Interestingly, the severity of anemia correlated with disease activity (ECLAM) only among patients with IDA but not among those with AIHA or ACD in the same report. Median time to remission with therapy is 3 months, and recurrence is unlikely in steroid responders; 85% are recurrence-free after 5 years and 73% remain so after 15 years of follow-up.1
Diagnosis
The diagnosis of AIHA is established in a step-wise fashion; a mechanistic algorithm is provided in eFig 34-3 and additional details in Box 34-2.
Box 34-2
Step-Wise Approach in the Diagnosis of Autoimmune Hemolytic Anemia in Systemic Lupus Erythematosus
Step 1: Is the Anemia Hemolytic?
In certain scenarios, haptoglobin or reticulocyte count may not reflect active hemolysis; haptoglobin is an acute phase reactant and may therefore be normal or even elevated, despite hemolysis in the presence of chronic inflammation or a co-existing tumor. Conversely, haptoglobin may be low in the absence of hemolysis in patients with chronic liver disease or with intramedullary hemolysis. A rare cause of low haptoglobin is congenital ahaptoglobinemia or hypohaptoglobinemia. The Hp0/Hp0 genotype is associated with undetectable serum haptoglobin and is relatively common in those of Korean descent (4%) but rare in Europe and North America (1:4000). Patients with the Hp2/Hp0 or Hp1/Hp0 genotype have low-serum haptoglobin.1,2 Likewise, reticulocytosis may be absent at diagnosis of hemolytic anemias. In one study, 25% of patients with proven hemolytic anemia had a normal reticulocyte count at presentation.3 The main reason may be a delayed bone marrow response; in the majority of patients, reticulocytosis can be demonstrated several days later. The absence of reticulocytosis may also be observed with reduced bone marrow function, such as autoimmune hemolytic anemia (AIHA) occurring during or after chemotherapy, and in patients with underlying infiltrating bone marrow disease similar to leukemia, lymphoma, or pure red cell aplasia (PRCA).
Step 2: Is it Autoimmune?
The significance of a positive or negative direct antiglobulin test (DAT) must be evaluated by the clinician. A positive DAT alone without active hemolysis is not sufficient for the diagnosis of AIHA, because 0.007% to 0.1% of the healthy population and 0.3% to 8% of patients who are hospitalized and without hemolytic anemia have a positive DAT result.5 In systemic lupus erythematosus (SLE), 18% to 65% of patients may have a positive DAT result without evident hemolysis.4 One reason for a false-positive DAT is hypergammaglobulinemia, for example, after high-dose immunoglobulin therapy. In the case of a negative DAT, the presence of immune hemolysis is unlikely but cannot be excluded. A false-negative DAT may occur in 1% to 10% of all patients with AIHA. A major reason, at least in the past, has been low sensitivity of the laboratory assay, which is no longer the case.5 Scenarios with a truly negative DAT include (1) patients with AIHA after treatment with rituximab who become DAT-negative, despite ongoing hemolysis; (2) AIHA in the context of chronic lymphocytic leukemia (CLL) after therapy with fludarabine, cyclophosphamide, or rituximab5; and (3) AIHA in the context of solid tumors.5
Step 3: What Antibody is Responsible for Aiha?
Cold antibody–AIHA (CA-AIHA) with high cold antibody titer (1:4096) and anti-I specificity has been reported only once in SLE.6 At large, the antibody is an IgM, measured by the “cold agglutinin assay,”5 and coated red cells are cleared by the liver. The DAT is positive with complement alone in 74% of patients with IgG and C3 in 22.4%, as well as with IgG alone in 3.4%. Both titer and thermal amplitude of cold agglutinin are clinically relevant. Patients with high titers and narrow amplitude have intermittent attacks of severe hemolysis. Conversely, hemolysis may occur at much lower titers when the thermal amplitude is high. CA-AIHA is generally characterized by extravascular hemolysis and considerably lower response rates to corticosteroids and cytotoxics, compared with IgG warm antibody-AIHA (WA-AIHA).
1 Park KU, Song J, Kim JQ. Haptoglobin genotypic distribution (including Hp0 allele) and associated serum haptoglobin concentrations in Koreans. J Clin Pathol. 2004;57(10):1094–1095.
2 Delanghe J, Langlois M, De Buyzere M. Congenital anhaptoglobinemia versus acquired hypohaptoglobinemia. Blood. 1998;91(9):3524.
3 Liesveld JL, Rowe JM, Lichtman MA. Variability of the erythropoietic response in autoimmune hemolytic anemia: analysis of 109 cases. Blood. 1987;69(3):820–826.
4 Voulgarelis M, Kokori SI, Ioannidis JP, et al. Anaemia in systemic lupus erythematosus: aetiological profile and the role of erythropoietin. Ann Rheum Dis. 2000;59(3):217–222.
5 Valent P, Lechner K. Diagnosis and treatment of autoimmune haemolytic anaemias in adults: a clinical review. Wien Klin Wochenschr. 2008;120(5-6):136–151.
6 Nair K, Pavithran K, Philip J, et al. Cold haemagglutinin disease in systemic lupus erythematosus. Yonsei Med J. 1997 Au;38(4):233–235.
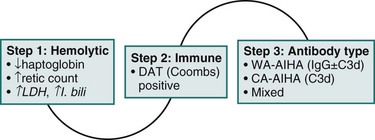
The second step is the differentiation between immune and nonimmune hemolysis. This is best predicated by the direct antiglobulin test (DAT), Coombs test. A positive test confirms the presence of bound antibodies (particularly IgG, but also immunoglobulin A [IgA] or immunoglobulin M [IgM]) and/or complement (C3d or C3c) on the surface of red cells through red cell precipitation, upon the addition of antihuman IgG antibody. A positive DAT in the context of established hemolytic anemia (step one) generally confirms the diagnosis of AIHA. The prognostic significance of antibody titers remains an unresolved question; patients with weak-positive DAT results may have severe hemolysis, whereas others with a strong-positive DAT result may be without overt anemia. In general, however, a strong-positive DAT result is more likely to be associated with severe AIHA.13
The third step is the identification of the type of antibody responsible for hemolysis. AIHA is classified into two major categories based on the optimal temperature of antierythrocyte antibody reactivity with antigens on the red cell surface. The warm antibody type (WA-AIHA) is mediated by antibodies optimally reacting with red blood cell (RBC) antigens at 37° C and causing hemolysis at 37° C. The cold antibody type (CA-AIHA) is mediated by IgM, a complement-fixing antibody that optimally binds RBC antigens at 4° C but mediates hemolysis at 37° C. SLE-associated AIHA is almost exclusively WA-AIHA.14 Conversely 6% of patients with WA-AIHA have SLE. RBCs coated by warm IgG undergo membrane alteration in vivo with each pass through the spleen, resulting in the formation of spherocytes; they are ultimately removed from circulation through phagocytosis, predominantly by splenic macrophages and, to a lesser extent, by sinus endothelial cells. The DAT is positive with IgG in 20% to 66% of patients with IgG plus complement (C3d) in 24% to 64% and with complement alone in 7% to 14% of all patients.
Two older studies reported AIHA in the presence of both warm IgG and cold IgM antibodies in the context of SLE. Sokol and colleagues15 found that 7% of 865 patients with AIHA had warm IgG and cold IgM anti–RBC antibodies, both of which contributed to hemolysis; of those two groups, 20% had SLE. Shulman and others16 reported that 5 of 12 patients (42%) with combined warm and cold AIHA had SLE. Patients with this type of AIHA have severe hemolysis but good response to corticosteroid therapy, similarly to IgG WA-AIHA.
Antigen Specificity of Antierythrocyte Antibodies
Antierythrocyte antibodies in SLE are mainly warm-type IgG, usually with non-rhesus specificity, otherwise undefined.1 In primary AIHA, such antibodies react with either Band 3 anion transporter protein on the RBC membrane or an epitope formed by Band 3 protein and glycophorin A. New Zealand black (NZB), lupus-prone mice produce anti-Band 3–specific antibodies.17 Interestingly, anti-Band 3 IgG antibodies are naturally formed in healthy patients, possibly functioning as eliminators of senescent erythrocytes, which, with aging, express Band 3 protein–derived neoantigens.18 The relationship between the naturally occurring and pathologic anti-Band 3 autoantibodies, as well as their differences in the context of SLE-associated AIHA, remains unknown.
(a) Anticardiolipin (aCL) IgG and IgM antibodies were highly prevalent (74%) in patients with SLE-associated AIHA, compared with both unselected patients with SLE19 and patients with SLE associated with IDA or ACD.1
(b) Mean IgG and IgM aCL titers were significantly higher in SLE-associated AIHA, compared with both unselected subjects with SLE and controls.20
(c) The presence of both APLA and AIHA in patients with SLE has been correlated with DAT positivity, as well as the reduction of complement receptor 1 (CR1) levels on the RBC surface that regulates C3 fragment deposition; both parallel disease activity.21
(d) Certain IgG and IgM APLA antibodies have been shown to bind erythrocytes and fix complement in vivo, accounting for the observed association of these antibodies with positive DAT results.22 The ability of this binding to decrease erythrocyte survival and cause AIHA was demonstrated in three patients with SLE; the antierythrocyte binding activity of eluted autoantibodies was totally inhibited by absorption with phospholipid micelles.
An interesting, yet unexplained, finding in patients with SLE-associated AIHA is an acquired deficiency in erythrocyte expression of CD55, or decay acceleration factor (DAF), and/or CD59, also known as a membrane inhibitor of reactive lysis (MIRL); compared to SLE subjects without AIHA, who may normally express such molecules. These are glycosylphosphatidylinositol-anchored membrane proteins, serving as complement regulators that protect erythrocyte membranes against complement activation and deposition leading to cell lysis. One study showed that the mean fluorescence intensity (MFI) of both CD55 and CD59 on erythrocytes of patients with SLE-associated AIHA is significantly reduced, compared with those without AIHA and with control patients.23 Interestingly, the underexpression of CD55 and CD59 has been reported on other formed elements in SLE and is likewise associated with autoimmune thrombocytopenia and lymphopenia.23 Since all three cytopenias do not always coexist in the same patients, a common mechanism affecting the expression in all three lineages is unsustainable. The fact that the underexpression of CD55 and CD59 may independently exist in different lineages insinuates the presence of lineage-specific pathophysiologic processes; not being an inherited defect, cell-specific antibodies appear as the most plausible alternative.23 However, the presence of anti-CD55 or anti-CD59 antibodies in SLE and their capacity to induce hemolysis has not yet been reported.
Treatment
General Considerations
(a) No established definitions of remission, complete or partial, are available. Certain reports have used benchmarks such as Hgb >12 g/dL and no hemolysis for complete remission; Hgb between 10 and 12 g/dL, or Hgb increase >2 g/dL, or hematocrit (Hct) >30% have been used as descriptors of partial remission. but without universal acceptance. Others have considered the reduction of reticulocyte counts, absence of hemolysis, or decreased need for transfusion as a benchmark for partial remission.
(b) No clinical trials address the efficacy and durability of first-line therapies (e.g., corticosteroids).
(c) No consensus of opinion is offered on when or what decides transition to second-line therapies (e.g., rituximab, splenectomy).
(d) No clinical trials exist that explore the effectiveness and longevity of second-line treatments.
(e) Especially in patients with SLE, AIHA frequently coexists with other manifestations such as nephritis, which may dominate therapeutic decisions. Consequently, most insight on the natural course and treatment of isolated AIHA in SLE is extrapolated from studies in idiopathic AIHA. As such, recommendations stem from retrospective studies, small series of (probably selected) patients or single cases (evidence level V), or a few prospective phase II trials, or are largely based on experience. As a general rule, AIHA occurs at the outset of SLE in two thirds of patients and generally recurs infrequently. The median time to remission with therapy is 3 months, 75% of patients respond to first-line therapy, 85% of responders are recurrence-free after 5 years, and 73% remain so after 15 years of follow-up.1
First-Line Therapies
For most cases of idiopathic and secondary AIHA including SLE, prednisone at a dose of 1 mg/kg/day is the first-line treatment of choice.24 Hgb response usually takes a few days; therefore some patients may require transfusion support. Transfusions should be avoided whenever possible, since patients with SLE develop isoantibodies against RBCs, as well as higher titers of isohemagglutinin antibodies more frequently than control patients. The efficacy of pretransfusion plasma exchange to decrease antierythrocyte antibody mass and to improve transfusion yield has not been validated.25 Approximately 80% to 90% of patients demonstrate a clear response (Hgb > 10.0 g/dL) within the first 3 weeks of treatment. Nonresponders, at that point, are unlikely to improve on steroids alone and should be considered for second-line therapy.26 After 3 months, two thirds are in complete remission and approximately 21% to 23% are in partial remission.27 Approximately 10% of all patients are nonresponders. When a response is achieved, the prednisone dose should be slowly tapered. Although not evidence-based, reducing the dose by 20 mg/day every 2 weeks down to a daily dose of 20 mg/day is generally recommended. In those who maintain response, the dose should be slowly reduced further by 5 mg or 2.5 mg/day every month. Considering the long half-life of erythrocytes, time is required to ascertain that a given dose of prednisone is sufficient to maintain Hgb within an acceptable target range, thus the reason for this strategy. Approximately 20% of adults remain in remission without further therapy, 40% to 50% require low-dose maintenance therapy, and 15% to 20% need a high maintenance dose of prednisone.
Second-Line Therapies
In patients with idiopathic AIHA, anti-CD20 monoclonal antibody (rituximab) and splenectomy are the only second-line therapies with proven short-term efficacy. In eight studies addressing rituximab in patients with AIHA at large, a clinical response was observed in 62 of 76 patients, based on the reduction in reticulocyte count, the absence of hemolysis, the decreased need for transfusion, or a normalization of Hgb.28 Similarly, in a Belgian retrospective study, 53 patients with primary and secondary AIHA were given rituximab after failing at least one previous therapy—including splenectomy in 19% of patients—and showed an overall response rate of 79% in a median follow-up of 15 months. Progression-free survival at 1 and 2 years were 72% and 56%, respectively.29 However, the reported experience with rituximab efficacy in adults with SLE-associated AIHA is limited (three case reports). As such, its use in SLE is reserved for severe or recalcitrant disease and perhaps patients with CA-AIHA or mixed WA-AIHA and CA-AIHA. The role of splenectomy is controversial in SLE-associated AIHA. Rivero and others30 compared the clinical course of 15 patients undergoing splenectomy for SLE-associated AIHA and/or immune thrombocytopenia and 15 SLE patients who were treated medically. Splenectomy produced short-term benefits, but, at follow-up, no difference between the two groups was observed. The splenectomy group had a significantly higher frequency of cutaneous vasculitis and serious infections after surgery. More patients undergoing splenectomy eventually required immunosuppressive therapy, compared with the medically treated group at follow-up.
Danazol in conjunction with corticosteroids has been reported as useful in WA-AIHA, including that associated with SLE.31 High-dose intravenous immunoglobulin (IVIG) has been used as a second-line agent after or concurrently with steroids but with low and transient efficacy. It was effective in 40 of 73 patients with WA-AIHA; thus IVIG is not recommended as a standard therapy but may be useful as an adjunct therapy for selected patients, such as those with toxicity to other treatments.32
Bone Marrow and Immune-Mediated Hematopoietic Failure in Systemic Lupus Erythematosus
The concept of hematopoietic failure as a result of an immune-mediated BM damage gained momentum in light of BM biopsy studies in patients with SLE-associated cytopenias. The largest study to date reported the histopathologic features in 40 patients with SLE and unexplained cytopenias from a single center33; hypocellularity, necrosis, and stromal changes such as edema and fibrosis along with vascular changes were frequently present.
BM was hypocellular in 58% of patients, normocellular in 17%, and hypercellular in 25%. Erythroid lineage was increased, normal, or decreased in 70%, 17%, and 13%, respectively, and myeloid lineage was increased in 13% and decreased in 17% of patients. Megakaryocytes were increased in 65% and decreased in 10% of patients.33 Dyserythropoiesis was uniformly observed and involved immature erythroid precursors; multinucleation, bizarre nuclear shapes, and budding were common, whereas nuclear karyorrhexis in erythroblasts was less prominent. Erythrophagocytosis was observed in 20% of patients. In addition, disruption of normal BM architecture was a prominent feature, affecting immature cells of all three lineages. In the normal human BM, myeloid precursors reside near the trabecular region, whereas erythroid and megakaryocytic precursors are located in the intertrabecular region. This distribution may be reversed in SLE, with erythroid and megakaryocytic precursors in the trabecular regions designated as abnormal localization of immature precursors (ALIP), which was present in 58% of patients and inversely correlated with Hgb (P = 0.01). These observations highlight the BM as a main target organ in SLE.
BM necrosis was present in 90% of patients with SLE and graded as mild in 58%, moderate in 22%, and severe in 10% of patients.33 Its morphologic features were distinctive—a smooth homogeneous basophilic background protein staining was often present. In addition, an increase in eosinophilic granular stroma was identified, along with the ghosts of many dead hematopoietic cells. The usual mechanism of BM necrosis is vascular obstruction, leading to ischemia. However, microvascular thrombosis or vasculitis was not seen, rendering the pathogenesis of BM necrosis unclear in patients with SLE. However, sinusoidal dilation and destruction of the lining endothelium was present in 20% of patients and was associated with the presence of moderate to severe necrosis (P = 0.008).33
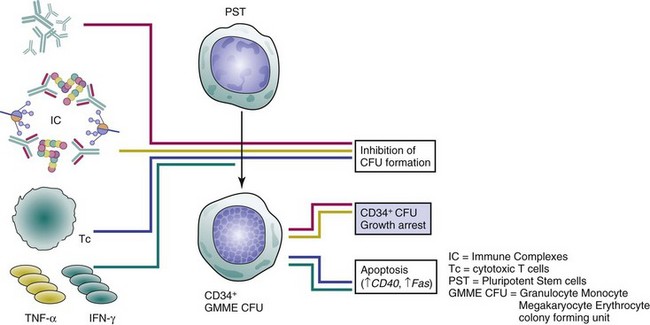
With regard to the immune system contribution to hematopoietic failure in SLE, evidence points to autoantibodies, immune complexes, and cytotoxic T cells as common effector mechanisms of progenitor growth arrest, inhibition of differentiation, apoptotic death, or BM stromal cell dysfunction (Figure 34-2). Complement-dependent or independent autoantibodies were found to suppress both erythroid- and granulocytic-colony formation by hematopoietic colony-forming units (CFU).34,35,36 IgG fractions of patients with active SLE cytopenias directly bound CD34+ CFU, causing growth arrest in vitro.37 However, no correlation was established between the severity of peripheral cytopenias and autoantibody inhibitory capacity, and the nature of respective antigen(s) remains elusive.37 Such antibodies culminate in syndromes similar to aplastic anemia, PRCA, myeloid hypoplasia, or amegakaryocytic thrombocytopenia and can be suppressed by immunomodulatory therapy.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


