79 Etiology and Pathogenesis of Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) represents one of the most significant diseases in all of medicine. Predominantly targeting young women in their childbearing years and with the potential to cause significant physical disfigurement, morbidity, and occasionally mortality, lupus is the focus of strong advocacy to support research that will generate insights into disease pathogenesis. In fact, characterization of the immunologic contributors to lupus, the prototype systemic autoimmune disease, has been the focus of particularly intense study since the flowering of the discipline of immunology in the 1950s and 1960s. Recent efforts to define the genetic variations that underlie susceptibility to lupus have supported the central role of the immune system in disease pathogenesis but have extended the view of lupus pathology beyond the important role of autoantibodies to include a significant contribution of the innate immune system to disease. An underlying role for the vasculature as a target of the immune system and its products is gaining renewed interest as an important component of lupus pathogenesis. Together, these recent advances provide important insights into how the intersection of genetic variations with environmental triggers amplifies immune system activation and target organ vulnerability to generate the classic manifestations of lupus and its clinically significant comorbidities. Figure 79-1 provides a schematic overview of many of the contributors to SLE and how they interact to drive autoimmunity and tissue damage.
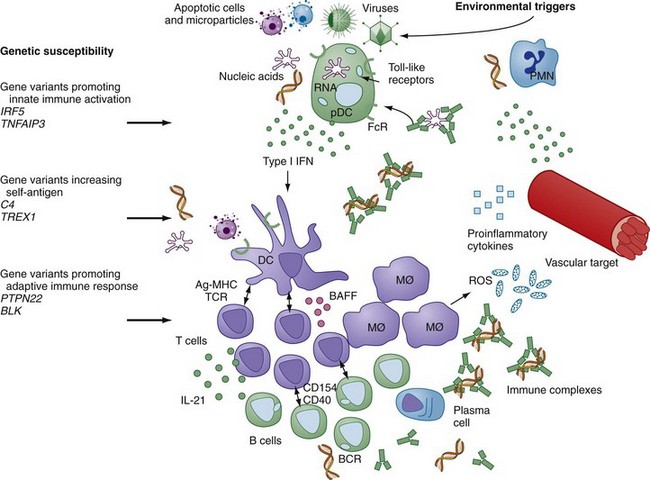
Genetic Contributions to Lupus Pathogenesis
Observations of several individuals with SLE within a family, along with a high frequency of concordance of SLE in identical twins, have pointed to a strong genetic contribution to SLE. Data suggest that concordance of clinical lupus disease in twins is 10 times more frequent in monozygotic than in dizygotic twins, although the highest reported concordance rate is still only 57%. Moreover, the suggestion that multiple autoimmune diseases are associated with common genetic susceptibility factors is supported by the aggregation of several distinct autoimmune diseases within a family. The pace of discovery of common genetic variants, typically identified by statistical analysis of single nucleotide polymorphisms (SNPs) in relation to a diagnosis of SLE, has markedly accelerated in recent years, as the cost of genotyping thousands of patient and control samples has become feasible. Publication of two important collaborative genome-wide association studies, one by the SLE Genetics (SLEGEN) consortium and the other organized by Genentech, identified at least nine new genes or genomic loci associated with SLE.1,2 Since presentation of those studies, additional lupus-associated genetic variants have been confirmed or at least strongly supported.3 The common theme among the lupus-associated genes is that the vast majority encode proteins implicated in immune system function. Although this result is not surprising, it confirms the essential contribution of the immune system to the autoimmunity, inflammation, and tissue damage that characterize lupus disease and points to the most significant molecular pathways that mediate altered immune function.
The lupus-associated genes that play likely roles in lupus pathogenesis can be grouped on the basis of their roles in immune function (Table 79-1). In parallel to the requirements for immune system activation stimulated by foreign antigens, SLE-associated genes are involved in generation of self-antigen, activation of the innate immune response, and activation of the adaptive immune response. Although the specific functional alterations that are conferred by the lupus-associated variant compared with the more common variant have yet to be defined in detail, in most cases there is sufficient information regarding function to allow formulation of hypotheses that can be tested in functional genetic studies.
Table 79-1 Genetic Variants Associated with Systemic Lupus Erythematosus (SLE)
GWAS, genome-wide association study.
The rare but high-risk deficiencies in complement pathway gene products, including C2, C4, and C1q, are thought to contribute to lupus pathogenesis by impairing clearance of cellular debris, a function that is typically supported by those complement components. Increased availability of nuclear debris can provide sufficient self-antigen for induction of self-reactive T cells or serve as an endogenous adjuvant for activation of the innate immune response. The ancestral major histocompatibility complex (MHC) 8.1 haplotype block, HLA-B8/DR3/DQw2/C4AQO, that is associated with lupus susceptibility, encodes the alleles B8 and DR3 and bears a short C4B gene and no C4A gene, whereas other nonrisk haplotypes carry either a longer C4B segment and/or one or more copies of C4A. In fact, the relative risk related to the C4A null allele is twice that of either HLA-B8 or DR3, pointing to the significance of the C4 genes in disease risk.4 It should be noted that this risk haplotype is also associated with accelerated disease in patients infected with human immunodeficiency virus (HIV), insulin-dependent diabetes mellitus, and several other autoimmune diseases. Deficiency of C1q, the recognition protein for the classical complement pathway, might contribute to disease on the basis of its important role in promoting clearance of apoptotic cell debris by mononuclear phagocytes.5 C1q plays an additional role in inhibition of IFN-α production by directing stimulatory immune complexes to monocytes rather than IFN-producing plasmacytoid dendritic cells. Through this mechanism C1q deficiency can augment IFN-α and promote broad immune dysregulation.6 C-reactive protein (CRP), a member of the pentraxin family, also contributes to clearance of apoptotic debris. Polymorphisms in CRP have been associated with SLE and with decreased levels of CRP, but it remains unclear how much of the genetic contribution to basal CRP levels is due to variations within the gene itself versus variations in other genes.
Recent studies of families with a lupus-like disease, the Aicardi-Goutières syndrome, suggest that mutations in genes encoding nucleases that cleave either DNA or ribonucleic acid (RNA) might result in excess stimulatory nucleic acid and innate immune system activation.7,8 Aicardi-Goutières and several related conditions are characterized by skin lesions, autoantibodies, central nervous system disease, and high levels of type I IFN. Mutations in the TREX1 gene, encoding DNase III, a 3′-5′ exonuclease, as well as two additional genes, RNASEH2 and SAMHD1, are associated with this syndrome.9 A functional connection between TREX1 and control of type I IFN production was demonstrated in a murine model in which TREX1 deficiency resulted in increased levels of IFN-β.10 A recent analysis of more than 8000 lupus patients found TREX1 mutations in approximately 0.5% of patients. In addition, a common variant in the TREX1 gene conferred an odds ratio for diagnosis of lupus of 1.73 compared with healthy controls.11 Taken together, these recent demonstrations of association of rare genetic variants of enzymes that regulate degradation of nucleic acids with SLE point to the central role of those nucleic acids as triggers for immune system activation and disease.
A large number of lupus-associated single nucleotide polymorphisms (SNPs) are found in genes that encode proteins involved in induction of type I IFN or response to that family of cytokines.12–14 IFN regulatory factor 5 (IRF5) and IRF7 are cytoplasmic proteins that translocate to the nucleus after effective activation of the endosomal TLRs by DNA or RNA. IRF5 and IRF7 then act as transcription factors to initiate transcription of IFN-α and other proinflammatory mediators. TNFAIP3 encodes A20, a protein that regulates several proinflammatory cellular mechanisms including signaling through endosomal TLRs and activation of nuclear factor κB (NFκB).15 Genetic variants in IRF5, IRF7, TNFAIP3, and numerous other lupus-associated genetic variants can be mapped to molecular pathways responsible for induction of innate immune system activation or responsiveness to its products, particularly IFN-α.
A third set of lupus-associated gene variants contributes to altered thresholds for lymphocyte activation or efficiency of cell activation. The ancestral MHC 8.1 haplotype that has been shown to be strongly associated with a diagnosis of SLE, along with other autoimmune diseases, appears to influence the early stages of immune system activation. Although more efficient presentation of self-antigens by disease-associated MHC class II alleles would seem to be the most likely mechanism by which the MHC risk haplotype confers predisposition to autoimmunity, available data point to a number of immune alterations, many focused on the T cell, in healthy individuals bearing the ancestral haplotype.4 The impact of those alleles is best defined clinically as determining immune responses that generate particular autoantibody specificities, and the alleles seem to be important in determining whether anti-DNA autoantibodies, antibodies specific for RNA-associated proteins, or both types of autoantibodies can be induced and produced through T cell–dependent B cell differentiation.16 Additional lupus-associated variants that alter adaptive immune system activation are involved in cytokine signaling such as STAT4 or efficiency of signaling downstream of the T and B cell surface antigen receptors such as PTPN22 in the case of both T and B cells and LYN, BANK, BLK, TNFAIP3, and others in the case of B cells.
A fourth category of lupus-associated genetic variants defines determinants of target organ damage. The understanding of genetic determinants of target organ vulnerability to immune mediated or oxidative damage is less well developed than is the role of genetic variants in altered immune system function in SLE, but identification of polymorphisms in members of the kallikrein gene family are associated with SLE and suggest areas for future investigation.17 Table 79-1 lists many of the genetic variants documented to have a statistical association with a diagnosis of SLE.
Although impressive progress based on GWAS has identified statistical association of sequence variations in genes or genetic loci with a diagnosis of SLE, the functional consequences of those variations have not been extensively characterized. The best developed insights regarding the impact of lupus-associated variants on immune function have derived from studies of gene products involved in production of type I IFN. The risk alleles for IRF5 and IRF7 are associated with increased serum type I IFN activity in those patients who demonstrate autoantibodies targeting DNA or RNA-associated proteins.12,18 Those studies support the hypothesis that nucleic acid–containing immune complexes are important stimuli that act through endosomal TLRs such as TLR7 and TLR9, those TLRs that mediate cell signals through the IRF5 and IRF7 transcription factors.
Subphenotype analysis will continue to amplify the information that can be gleaned from patient and control genotyping. A recent study of anti-dsDNA+ and anti-dsDNA− SLE patients, along with healthy controls, identified HLA-DR3, STAT4, and ITGAM as significantly associated with the presence of anti-dsDNA antibodies.19 A case-only analysis additionally associated PTPN22, IRF5, and PTTG1 with anti-dsDNA antibodies. In contrast, a distinct group of lupus-associated genes showed comparable association between anti-dsDNA positive and negative patients, including FCGR2A, OX40L, IL10, PXK, UHRF1BP1, PRDM1, BLK, and IRAK1. Although it is possible that some of these variants are associated with distinct autoantibody specificities, it is more likely that they represent risks for other aspects of lupus pathogenesis, perhaps contributing to inflammation and tissue damage.
Whether the insights regarding specific genetic susceptibility factors can be used to predict development of lupus or particular manifestations of disease is as yet unclear. Recent studies have attempted to determine the predictive value of accumulated genetic risk variants. Although increased numbers of lupus-associated alleles are associated with some increased risk of disease and the number of lupus-associated genetic alleles is significantly higher in patients with anti-dsDNA antibodies than in those without those autoantibodies, at this time, it would not appear that genotyping for lupus risk is sufficiently informative to warrant practical application in patient management.20
Female Predominance of Systemic Lupus Erythematosus
A discussion of genetic contributions to SLE pathogenesis cannot ignore the dramatic 9 : 1 female predominance of the disease. Of all of the characteristic clinical features of lupus, it is the extreme sex skewing that remains least understood. Hormonal contributions to immune system activation are likely to represent a component of the female predominance of the disease; estrogen can modulate lymphocyte activation, and prolactin has been shown to be expressed at increased levels in lupus serum.21
Granting a contribution of hormones to increased immune activation, it would appear that additional concepts should be entertained to understand why 9 or 10 females are diagnosed with SLE for every male lupus patient. It is intriguing that Klinefelter’s syndrome, characterized by a 47,XXY genotype, is increased 14-fold among men with SLE compared with men without SLE.22 These data are proposed to support an X chromosome gene dose effect as an important contributor to SLE pathogenesis. Identifying the X chromosome as a possible risk factor for SLE provides a basis for new hypotheses regarding disease susceptibility, but the nature of that risk remains undefined. A possible role for altered regulation of epigenetic processes such as DNA methylation has been raised, and murine studies have supported the impact of duplications of portions of the X chromosome that encode the TLR7 gene in activation of the innate immune system, production of type I IFN, and generation of autoimmunity.23 Investigation of altered gene dosage in human patients has not confirmed a similar duplication of the TLR7 gene, and studies of DNA methylation are most suggestive of altered DNA methylation reflecting generalized immune activation rather than a primary etiologic event.24 Further studies of epigenetic regulation of X chromosome structure and gene expression are warranted.
A potential pathogenic role for the distinct events that occur in the ovary compared with those in the testes also deserves further study. Onset of SLE most typically occurs in the childbearing years, after menarche and before menopause. A positive association between early menarche and SLE has been observed, and breastfeeding confers a protective effect.25 Both observations might be consistent with molecular and cellular events related to ovulation as factors contributing to lupus pathogenesis. It is intriguing to consider that the biology of germ cell maturation, with female germ cells undergoing a second meiotic division before ovulation each month, might involve mechanisms and mediators that affect immune recognition. Although not yet fully understood, the carefully orchestrated demethylation and remethylation of DNA, along with the production of regulatory RNAs and RNA-associated proteins such as so-called PIWI proteins, in the germ cells and associated somatic cells might provide a source of stimulatory nucleic acid–containing complexes that could access TLR-dependent or TLR-independent pathways and result in immune activation.26 The challenge in pursuing such concepts is the obvious limitation on access to ovarian tissue for study, although murine models might be helpful in that regard.




