CHAPTER 31 Congenital Anomalies of the Spinal Cord
Myelomeningocele
Myelomeningocele is the most common significant birth defect involving the spine. The condition is manifest at birth and is characterized by herniation of a malformed spinal cord through a defect in the bony canal and skin. It almost always results in permanent disability regardless of medical intervention and often deprives the victim of “those qualities held in high esteem by our society—independence, physical powers and intelligence.”1 In the past, it was assumed that all of the neurologic dysfunction that occurs with myelomeningocele arose from disordered embryogenesis. More recent work suggests, however, that at least some of the spinal cord damage is acquired in utero,2 and fetal surgery to close the spinal defect is now being performed at selected centers.
Embryology
By 18 days of development, the embryo is a flattened oval disc with all three germ layers present. A longitudinal depression, the neural groove, appears in the neural plate, which is destined to become the brain and spinal cord. By 22 days, the neural groove has deepened, and fusion of the adjacent tissue begins the transformation of the flat neural plate into a hollow neural tube. The entire process is called neurulation, which begins in the dorsal midline and simultaneously progresses cephalad and caudad. The final portions of the tube to close are the rostral opening (the anterior neuropore, at 24 days) and the caudal opening (the posterior neuropore, at 28 days). By the 1st month of gestation, the entire process has been completed. The development of the meninges begins after closure of the posterior neuropore, as does formation of the bony laminae.3
Myelomeningocele presumably occurs when the posterior neuropore fails to close or if it reopens as the result of distention of the central canal of the spinal cord with cerebrospinal fluid (CSF). The spinal abnormality is only one component of a complex of central nervous system abnormalities, which include the Chiari II malformation, hydrocephalus, and brain anomalies such as partial agenesis of the corpus callosum and gyral malformations. McLone and Naidich4 attempted to explain these associations with their “unified theory.” It is hypothesized that the open spinal defect allows excessive drainage of CSF in utero and that this results in collapse of the rhombencephalic vesicle, resulting in a small posterior fossa volume. Growth of the cerebellum and brainstem within a small posterior fossa results in downward herniation and caudal displacement of the cerebellar vermis and brainstem into the cervical spinal canal (the Chiari II malformation). Because the outlet of the fourth ventricle is occluded by impacted brain tissue, obstructive hydrocephalus develops either in the fetal period or in the newborn period after closure of the myelomeningocele eliminates the spinal defect as a drainage pathway.
Epidemiology
The incidence of myelomeningocele ranges from less than 1 case per 1000 live births in the United States to almost 9 cases per 1000 in areas of Ireland. The etiology of myelodysplasia is unknown, and evidence exists for environmental and multifactorial genetic influences. A role for genetic risk factors is supported by numerous studies documenting familial aggregation of this condition. In addition, several lines of evidence point to the potential importance of maternal nutritional status as a determinant of the risk for having a child with spina bifida. Indirect support is provided by studies that indicate that season of conception, socioeconomic status, and degree of urbanization may be related to the risk of spina bifida. Several micronutrients (vitamins C and B12, zinc, and folic acid) have been implicated as potential risk factors as well.5
Folic acid is the most extensively studied micronutrient. In August 1991 after the Medical Research Council Vitamin Study Group report was published, the U.S. Centers for Disease Control and Prevention (CDC) advised that women with a history of an affected pregnancy should take 4 mg of folic acid daily, starting at the time they planned to become pregnant.6 A dose of 0.4 mg was later recommended for all women of childbearing age capable of becoming pregnant. It was anticipated that these recommendations would have a substantial impact on the risk of neural tube defects in the offspring of such women. Most affected pregnancies (approximately 95%) occur in women with no history of a prior affected fetus or child, however.7 It has been suggested that fortification of the food supply may be more effective than individual supplementation for the prevention of neural tube defects.
Prenatal Diagnosis
More recent developments in prenatal diagnosis of fetal anomalies have made antenatal recognition of myelomeningocele commonplace. Families at risk are routinely offered amniocentesis for amniotic α-fetoprotein and acetylcholinesterase, which are important in separating open lesions from skin-covered masses such as myelocystocele. Screening ultrasonography performed on mothers when the fetus is at 16 to 20 weeks of gestation has also proved valuable.8 Other anomalies may be detected, and ultrasonography may detect skin-covered lesions such as lipomyelomeningocele. The cardinal finding is splaying of the posterior elements of the spine in the axial plane in the lumbosacral region. Indirect signs are the lemon sign, which refers to a bilateral concave contour of the frontal bones of the skull, and the banana sign, which describes anterior curvature of the cerebellar hemispheres. Hydrocephalus is also readily detected. In referral centers, diagnostic sensitivity is close to 100% in diagnosing spina bifida.9
Prenatal magnetic resonance imaging (MRI), using ultrafast T2-weighted sequences, may also be used to characterize the Chiari II malformation and other associated anomalies.10 Fetal MRI may detect spinal cord abnormalities in instances where ultrasonography can detect only bony abnormalities.11 Studies indicate that such prenatal imaging studies can help to determine prognosis. Specifically, lesion level determined by prenatal imaging studies seems to predict neurologic deficit and ambulatory potential, but the degree of fetal ventriculomegaly and the extent of hindbrain deformity are not predictive.12 Families can be professionally counseled regarding the expected prognosis and offered conventional treatment, abortion, or the possibility of fetal closure. Patients of low socioeconomic class, who are most at risk for neural tube defects, often do not present for prenatal care until after 24 weeks of gestation, when screening is no longer of value.
The value of prenatal diagnosis of spinal dysraphism is that an opportunity is provided to terminate the pregnancy, to evaluate the fetus for possible fetal surgery, and to determine the best mode of delivery. It has been proposed that children with myelomeningocele sustain traumatic damage to the placode and nerves when they are delivered vaginally and that children electively delivered by cesarean section before the onset of labor have improved motor levels.13 Other investigators have found little benefit to this strategy,12 but in the United States cesarean delivery has become routine.
Initial Evaluation
The initial assessment of a newborn infant with a myelomeningocele begins with a detailed examination to evaluate general well-being and to seek associated anomalies. Fatal urologic or cardiac anomalies may be evident that would favor nonoperative treatment. Some infants may have abnormal facies suggesting Down syndrome, and although chromosomal studies should be obtained, these are most often normal, and the facial appearance becomes normal with age. Approximately 85% of infants with myelomeningocele present with hydrocephalus or develop it within the newborn period.14 A large head circumference or bulging fontanel suggests the need for early head ultrasonography via the fontanel. Stridor, apnea, or bradycardia in the absence of overt intracranial hypertension suggests a symptomatic Chiari II malformation and hindbrain dysfunction, which carries a poor prognosis.
The neurologic examination is difficult in neonates, and it is easy to mistake reflex motion for voluntary movement. Fixed contractures and foot deformity suggest paralysis of the spinal segments innervating the joints. Any movement in response to painful stimulation of the same extremity must be viewed as potentially reflexive. Crying in response to a painful stimulus suggests intact sensation at that level. Table 31–1 lists the segmental innervation of the lower extremities and may be used as a guide in assigning a functional level. It is best, however, to document function of the individual muscle groups rather than simply to record a spinal level.
TABLE 31–1 Innervation of Leg Muscles
| Hip flexion | L1-3 |
| Hip adduction | L2-4 |
| Knee extension | L2-4 |
| Ankle inversion | L4 |
| Toe extension | L5-S1 |
| Hip abduction | L5-S1 |
| Hip extension | L5-S1 |
| Knee flexion | L5-S2 |
| Ankle plantar flexion | S1-2 |
Data from Sharrard WJW: The segmental innervation of the lower limb muscles in man. Ann R Coll Surg 35:106-122, 1964.
Management of an Unrepaired Newborn
Pending plans for definitive care, the infant is nursed in the prone position with a sterile saline-soaked gauze dressing loosely applied to the sac or placode. Broad-spectrum antibiotics (ampicillin and cefotaxime) are begun intravenously pending discussion with the parents. If there is no sign of overt hydrocephalus, the back is closed initially, and hydrocephalus is treated with a ventriculoperitoneal shunt at a separate procedure if needed. In patients with hydrocephalus and intracranial hypertension at birth, it may be advisable to perform both procedures at the same time because failure to treat the hydrocephalus may allow continued leakage of CSF into the back and threaten the closure and because the infant is exposed to a single anesthetic. The goal of back closure is to seal, using multiple tissue layers, the spinal cord and subarachnoid space against entry of bacteria from the skin. At the same time, the surgeon must preserve whatever neurologic function remains and attempt to prevent tethering of the spinal cord. The cross-sectional anatomy of a typical myelomeningocele is shown in Figure 31–1.
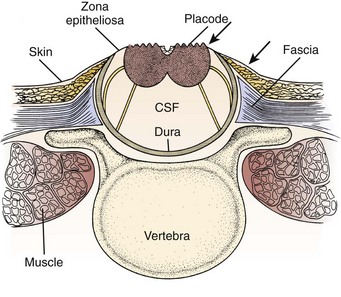
FIGURE 31–1 Cross-sectional anatomy of typical lumbosacral myelomeningocele. CSF, cerebrospinal fluid.
The infant is positioned in the prone position under general anesthesia. Rolls are placed under the chest and hips to allow the abdomen to hang freely and minimize epidural bleeding (Fig. 31–2A). If the sac is intact, fluid is aspirated and sent for culture. The surgeon gently attempts to approximate the base of the sac or defect vertically and then horizontally to determine which direction would produce the smallest skin defect. An elliptical incision is made, oriented along that axis, outside the junction of the normal, full-thickness skin and the thin, pearly zona epithelioserosa. Full-thickness skin forming the base of the sac is viable and should not be excised. This incision is carried through the subcutaneous tissue until the glistening layer of everted dura or fascia is encountered. The base of the sac is mobilized medially until it is seen to enter the fascial defect (Fig. 31–2B).
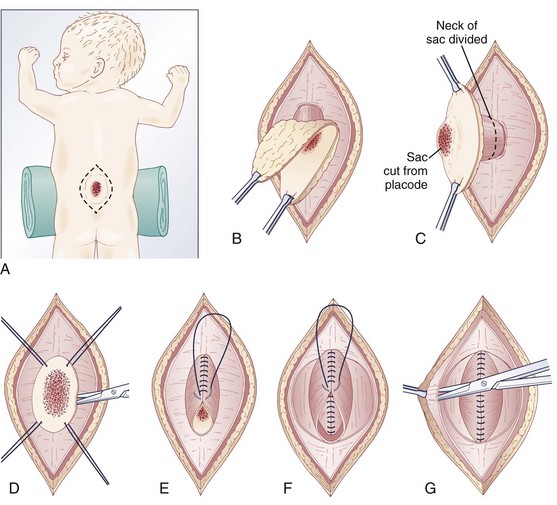
The sac is entered by radially incising the cuff of skin surrounding the placode. This skin is sharply excised circumferentially around the placode and discarded, with care being taken to avoid damaging the placode (Fig. 31–2C). All of the zona epithelioserosa is removed to prevent later development of an epidermoid cyst. At this point, the placode is floating freely inside the everted dura (Fig. 31–2D). In some thoracic myelomeningoceles, the placode is large and “thinned out,” and there is complete paraplegia below the level of the defect. In these instances, it may be appropriate to excise the placode to prevent spasticity and high-pressure bladder dysfunction.
Attention is now directed toward the dura, which is everted and loosely attached to the underlying fascia. It is undermined and reflected medially on each side until enough has been mobilized to effect closure (Fig. 31–2E). The dura is very thin anteriorly where the root sleeves exit and is easily torn. When it is free, the dura is closed in a watertight fashion with 4-0 nonabsorbable suture material.
Particularly if the dural closure is suboptimal, it is desirable also to close the fascia as a separate layer. The fascia is incised laterally in a semicircular fashion on either side, elevated from the underlying muscle, and reflected medially (Fig. 31–2F). It is closed with 4-0 suture over the underlying dural closure. The fascia is poor at the caudal end of a lumbar myelomeningocele or with sacral lesions, and the closure may be incomplete.
Mobilization of the skin is by blunt dissection with scissors or a finger; it may be necessary to free it anteriorly to the abdomen (Fig. 31–2G). In most instances, the closure is easiest in the mid-sagittal (vertical) plane, but occasionally less tension is required for a horizontal closure. A two-layer closure is performed. The epidermis may be closed with absorbable suture material and tissue glue if there is little tension, but interrupted nonabsorbable suture material is preferred for large defects.
Very large lesions require special techniques. There is often an associated gibbous deformity, and it is helpful to use a rongeur to remove the everted lamina, which, if left in place, would produce pressure points on the skin closure. Large circular defects may be closed by an S-shaped skin opening, allowing the use of rotation flaps (Fig. 31–3). Alternatively, a Z-rhomboid flap, tissue expanders, or latissimus dorsi myocutaneous flaps may be employed. Lateral relaxing incisions with split-thickness skin grafting have been described, but should be avoided if possible because the cosmetic result is poor. Alternatively, allogeneic skin grafts such as Alloderm (Lifecell, Branchburg, NJ) can be sewn directly to the edges of an approximated closure and may offer a better cosmetic result.15 In rare cases where patients are referred late for surgery, significant scarring of the placode can occur. Tissue expansion is particularly useful in such cases.16
Large defects occasionally undergo dehiscence, owing to tension on the skin closure or superficial infection. This dehiscence is usually evident by 7 days postoperatively. In most cases, the underlying fascial and dural closures remain intact. The dehiscence may be treated by removing the sutures to the point of good skin closure, débriding the devitalized tissue, and beginning a program of saline wet-to-dry dressings. When a good granulation bed is observed, the wound can simply be allowed to granulate and contract or may be secondarily closed or repaired with a skin graft with or without tissue expansion.17 For persistent wound breakdown, the pedicled “propeller” flap can be used as a vascularized salvage procedure.18 CSF leakage is treated by a ventricular diversion procedure, even if the ventricles are small.
Closure Before Birth
In a few centers, a fetus with spina bifida may be a candidate for in utero treatment because this condition is routinely detected before 20 weeks of gestation (Fig. 31–4). There is evidence that neurologic deterioration occurs during gestation. Normal lower extremity movement can be seen on ultrasound studies of affected fetuses before 17 and 20 weeks of gestation, whereas most late-gestation fetuses and newborns have some degree of deformity and paralysis. Such deterioration could be the result of exposure of neural tissue to amniotic fluid and meconium or direct trauma as the exposed neural placode impacts against the uterine wall. Theoretically, such deterioration could be reduced or eliminated by in utero closure of the lesion.
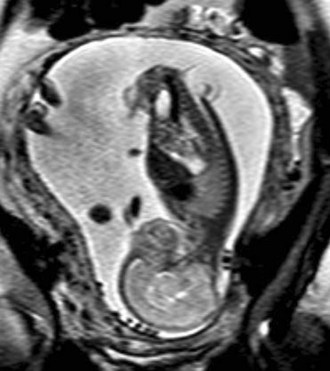
FIGURE 31–4 Fast spin-echo T2-weighted fetal MRI of a 20-week fetus with myelomeningocele. The sac is intact.
Some animal studies (in which a model for spina bifida is created by laminectomy and exposure of the fetal spinal cord to the amniotic fluid) have shown improved leg function if the lesion is closed before birth.19 Although other animal studies have failed to show behavioral improvement, they have documented the presence of somatosensory evoked potentials (SSEPs) in the treated myelomeningocele group compared with an absence of SSEPs in the untreated group.20 There is also evidence that the Chiari II malformation, which occurs in most individuals with spina bifida, is acquired and could potentially be prevented by in utero closure.21
In 1997, in utero repair of spina bifida was performed by hysterotomy at Vanderbilt University and at The Children’s Hospital of Philadelphia.22,23 Fetuses treated in utero were subsequently delivered by cesarean section, ideally at around 36 weeks of gestation. The early experience at both institutions suggested that compared with infants treated postnatally, fetuses treated in utero had a decreased incidence of hindbrain herniation and possibly a decreased need for shunting.24,25 The combined experience at the Children’s Hospital of Philadelphia and Vanderbilt University indicates that the incidence of hydrocephalus requiring shunting in patients treated in utero is less than that of historical controls stratified by spinal level who received standard postnatal care.26,27 It is hypothesized that fetal closure of the spinal lesion reduces the need for shunting by eliminating the leakage of CSF, which puts back-pressure on the hindbrain. The hindbrain hernia can be reduced, and the obstruction of the outflow from the fourth ventricle is relieved.28
It is now estimated that more than 330 in utero spina bifida closures have been performed.29 The procedure seems to be generally well tolerated by the expectant mothers. The fetal death rate is about 5% and is due to uncontrollable labor and premature birth. One analysis of leg function in children treated prenatally revealed no significant difference from a set of historical controls treated with conventional postnatal repair. Many of the children evaluated in this series had lower limb paralysis at the time of the surgery, however, which may have diluted any possible benefit.30 Data from the Children’s Hospital of Philadelphia suggested possibly better leg function in patients who already had intact leg movement shown by ultrasound before fetal sugery.31 Problems with delayed development of dermoid inclusion cysts and tethered cord may adversely affect long-term outcome.32 The preliminary experience suggests that children treated in utero have the same urodynamic abnormalities that are seen in conventionally treated children with spina bifida.33,34
To date, outcomes for spina bifida infants treated in utero have been assessed relative to outcomes in conventionally treated, historical controls.14 Such comparisons are prone to substantial bias, however, because fetuses who undergo in utero closure represent a highly selected subset of cases. In addition, the medical management of spina bifida is continuously improving, making comparisons with historical controls particularly problematic. Definitive answers about the benefits of fetal closure can be obtained only by a properly designed and conducted randomized trial.
Surgical Complications
Latex Allergy
An increased incidence of allergy to latex, as found in surgical gloves, balloons, and urinary catheters, has been well documented in children with spinal dysraphism.35 Allergy to latex is a type I, immediate, IgE-mediated reaction that can lead to anaphylaxis and death. Sensitization presumably arises from repeated exposure, at home and in the operating room. In many centers, all children with spina bifida are managed in a latex-free environment, and surgical procedures are preceded by prophylactic medications.
Late Deterioration
Tethered Cord
Approximately 20% to 30% of patients with myelomeningocele experience spinal cord tethering, which may result in progressive loss of function similar to that seen with occult spinal dysraphism.36,37 This phenomenon does not seem to be reduced by fetal repair, in which inclusion cysts have been found in 19% of patients who underwent fetal repair.38 Signs and symptoms usually appear during the 1st decade of life when growth is rapid and include back and leg pain, decrease in urinary control, gait difficulty and leg weakness, hip dislocation, progressive foot deformity, and possibly scoliosis. Urinary and gait difficulties are complaints confined to patients with low-level lesions, who are already functioning quite well neurologically and have the most to lose. In more severely affected patients who may already be wheelchair bound, pain may be the only manifestation of cord tethering and may be confused with appendicitis, hernia, urinary tract infection, or traumatic arthritis associated with abnormal posture or gait.
As with lipomyelomeningocele, the aim of surgery is to prevent further deterioration of function, but occasionally improvement is seen, even in preexisting deficits of long-standing duration. Reigel39 reported improvement in bladder function in five of nine patients with recent deterioration and in 77% of patients with gait difficulty and motor weakness. Pain almost invariably is relieved. Retethering can occur, and if new symptoms suggest this as an etiology, reoperation is indicated. An evidence-based review of the literature concluded that aggressive tethered cord release should be performed in adults within 5 years of symptom onset.40 Post–myelomeningocele repair patients tend to fare worse, however, than adult patients with closed dysraphism.
Hydromyelia
The routine availability of MRI has heightened awareness of this entity, which may be found in 68% of myelomeningocele patients.36 Hydromyelia presumably is the result of hydrodynamic forces arising from persistent fetal or untreated hydrocephalus that forces CSF down the central canal of the spinal cord.4 This explanation accounts for the frequency of symptomatic hydromyelia in patients with unshunted ventriculomegaly and patients considered to have “compensated” hydrocephalus, whose nonfunctional shunts were not revised because they lacked overt signs or symptoms of intracranial hypertension.41 An alternative explanation has been proposed by Oldfield and colleagues,42 who used dynamic MRI to study CSF flow. They suggested that a systolic pressure wave in the cranial compartment is transmitted in a pistonlike manner to the cerebellar tonsils, causing a systolic spinal CSF pressure wave that pushes CSF into the cord through its outer surface.
Symptoms of hydromyelia in spina bifida differ from symptoms in classic syringomyelia. The latter entity typically produces a dissociated sensory loss (relative loss of pain and temperature modalities with preservation of light touch), atrophy, and fasciculations primarily affecting the shoulder girdle and hands. When found in association with myelomeningocele, hydromyelia typically results in progressive bladder dysfunction, spasticity of upper and lower extremities, quadriparesis, and generally preserved sensation. The myelomeningocele repair may become swollen and tender. Symptoms may be due in part to the direct effects of the hydrocephalus stretching the cortical motor fibers. Hydromyelia may also be seen with progressive scoliosis. Spinal cord tethering may be the primary underlying etiology, however, of scoliosis and hydromyelia in these cases.43
Workup consists of MRI of the brain, followed by MRI of the entire spine to visualize the extent of the hydromyelia and the associated Chiari malformation and tethered spinal cord (Fig. 31–5). The hydromyelia cavity may be localized to a few spinal segments or extend throughout the length of the spinal cord. There may be multiple cavities and septations.
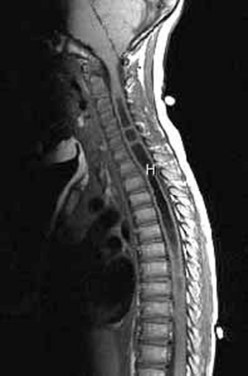
FIGURE 31–5 T1-weighted mid-sagittal MRI of a patient with Chiari malformation and large septated hydromyelia (H).
Other procedures have been described. Terminal ventriculostomy was advocated by Gardner and colleagues44 for classic syringomyelia, but the myelotomy tends to scar closed with time. In addition, the negative pressure provided by the pleural end of a shunt provides more decompression than simply allowing the hydromyelia to communicate with the subarachnoid space. Park and colleagues45 advocated posterior fossa decompression and plugging of the obex to prevent CSF fluid from the fourth ventricle from entering the central canal in patients in whom ventriculoperitoneal shunt revisions do not alleviate symptoms. This is a formidable procedure, however, and recurrent vomiting may occur from irritation or compression of the brainstem at the obex.
Chiari Malformation
MRI of the brain and spine is the procedure of choice for initial evaluation because the lesion is readily visualized and associated abnormalities are easily excluded. Mid-sagittal views show the tongue of cerebellum extending below the foramen magnum in type I lesions (see Fig. 31–5) and the “vermian pseudotumor,” consisting of the fused brainstem and cerebellum, in type II lesions.
Chiari type II lesions manifesting in an infant with myelomeningocele are particularly difficult to manage. When symptoms are severe, the brainstem nuclei may be irreversibly damaged, and the patient would not improve with surgical decompression.46 This situation has prompted a trend toward earlier intervention, when improvement may still be possible.48 If shunt function is shown to be optimal with shunt tap or imaging, Chiari decompression should be undertaken.
Patients are operated on in the prone position. A midline incision is used to expose the suboccipital bone and the cervical spine down to the level of the cerebellar hernia as seen on MRI, and a laminectomy is performed. The tonsillar hernia can usually be seen through the translucent dura, which is opened in the midline, beginning at the lowest point of the laminectomy and progressing cephalad. In this way, MRI-compatible clips or a running suture can be used to control bleeding from the anomalous venous sinuses in the posterior fossa. The dural opening should include the fibrous band usually present at the foramen magnum; however, because the foramen magnum is typically large and the venous sinuses are caudally displaced, the dural incision should rarely be carried above the level of the foramen magnum. It may be possible to open the fourth ventricle and establish CSF flow to the subarachnoid space, but dense adhesions and distorted anatomy often may make this unwise. As in Chiari I repair, the dura is left open, or a dural graft is employed, and the muscles, fascia, and skin are closed as usual.49
Shunt Malfunction and Arrested Hydrocephalus
Any neurologic deterioration in a patient with myelomeningocele should prompt a thorough investigation of the shunt. Some patients have nonfunctioning shunts and ventriculomegaly yet have no overt symptoms of intracranial hypertension or may have never received shunts and have baseline ventriculomegaly. These patients have been described as having “compensated” or “arrested” hydrocephalus. It has been suggested that some of these patients might benefit from shunting or shunt revision, which in some instances may result in improved neuropsychological functioning.50
Outcome
Significant progress has been made in the understanding and management of myelomeningocele over the past 25 years, particularly in the widespread use of multidisciplinary teams of specialists to manage children with this condition. More recent population-based data indicate that 1-year survival of individuals with spina bifida is approximately 92%51 and that 78% of all individuals with spina bifida survive to age 17 years.52 Mortality continues into the adult years53: 57% of patients die by the 4th decade of life.54 Death is due to problems associated with the Chiari II malformation, restrictive lung disease secondary to chest deformity, shunt malfunction, and urinary sepsis. Perineal sensation and urinary continence may be predictive of survival.55
Approximately 75% of young children with myelomeningocele are ambulatory to some extent, although most require braces and crutches. The likelihood that a child will ambulate is related to the level of the lesion; virtually all patients with sacral and lumbosacral lesions walk, but only half of patients with thoracic or thoracolumbar lesions walk with the use of braces and crutches. Patients with high-level lesions often can walk as young children but become wheelchair users as they get older, gain weight, or simply discover that wheelchair locomotion requires less energy and is faster than ambulation with crutches. A more recent report compared ambulatory potential between patients delivered by cesarean section and patients who underwent a trial of labor. Compared with the elective cesarean group, patients in the trial of labor group were more likely to be ambulatory at 2 years (independently ambulant 7% vs. 28%, ambulant with assistance 63% vs. 65%, or wheelchair bound 30% vs. 7%) and at 10 years (independently ambulant 5% vs. 21%, ambulant with assistance 30% vs. 54%, or wheelchair bound 65% vs. 25%). There was no statistical difference based on mode of delivery.56
Overall, approximately 75% of surviving infants have normal intelligence (IQ >80). This percentage decreases slightly to approximately 70% for surviving adults.57 Numerous factors predict poor intellectual outcome. In one study, the mean IQ of infants who were not shunted was 104; of infants shunted without complications, it was 91; and of infants shunted who had complications such as ventriculitis or anoxia, it was 70.58 Intelligence is also related to the level of the lesion: Approximately 55% of infants with thoracic lesions have significant developmental delay compared with only 25% of infants with lesions at lower levels.59
The cause of mental retardation in children with myelodysplasia remains controversial. McLone and colleagues60 attributed it to ventriculitis, but this does not account for all cases, and it is likely that in most cases forebrain dysfunction is simply part of the complex of anomalies associated with myelodysplasia. With respect to fetal surgery, preliminary evidence from the Children’s Hospital of Philadelphia showed that two thirds of surviving patients are within normal cognitive range at 2 years of age.27 Because cognitive function seems to be related to shunting, it is hoped that a decrease in overall shunting rate may improve overall intelligence in this population.
Although virtually all children with myelomeningocele have abnormal bladder function, urinary continence with the use of clean intermittent catheterization approaches 90% in children 5 to 9 years old. Ongoing urologic care is essential.61 Scoliosis is present in 49%, with 43% eventually requiring spinal fusion, for which the procedural complication rate is extremely high.62 Of children, 23% have at least one seizure. A tethered cord release is performed in 32%, with 97% having improvement or stability of preoperative symptoms.53
Occult Spinal Dysraphism and Tethered Cord
Embryology
After closure of the neural tube, the fetal spine is covered by ectoderm, and the low lumbar and sacrococcygeal segments have not developed.3 At this point, the caudal end of the neural tube blends with a large aggregate of undifferentiated cells, the caudal cell mass. A series of vacuoles in this mass coalesce and achieve continuity with the central canal of the previously formed neural tube (at roughly 29 days’ gestation), a process called canalization. The third phase involves retrogressive differentiation, during which the previously formed tail structures undergo a precise, ordered necrosis, leaving only the filum terminale, the coccygeal ligament, and the terminal ventricle of the conus as remnants by 11 weeks. Cell rests with potential for differentiation may be left within these structures, accounting for the development of lipomas, hamartomas, ectopic renal tissue, and the rare malignancy occasionally found in association with occult spinal dysraphism. Failure of the caudal cell mass to regress presumably gives rise to the hypertrophied filum terminale.
Pathogenesis
Symptoms may be caused in several ways. First, abnormal formation of the spinal cord and roots during embryogenesis may result in permanent deficits manifest at birth, as seen in myelomeningocele. Second, local masses growing within the rigid bony spinal canal may compress the conus medullaris or cauda equina and cause mechanical distortion or ischemia, with consequent dysfunction of these neural elements in a progressive fashion.63 This mechanical distortion may account for the acceleration of symptoms seen in some patients with significant weight gain in whom a lipoma enlarges in proportion to other body fat stores.
Finally, symptoms may be produced by traction on the spinal cord. Early in embryonic development, there is a progressive and rapid ascent of the conus within the bony spinal canal owing to faster growth of the vertebral bodies compared with the spinal cord. This “ascent of the conus” in children is slight, being only one segment, from L3 to L2 in the period from the 26th week of intrauterine life until maturity.64 Nonetheless, if the conus is tethered to the bony spinal canal, as often occurs in dysraphism, there is loss of mobility of the conus during spinal flexion and extension. Experimentally, spinal cord tethering has been shown to interfere reversibly with spinal cord oxidative metabolism65 and is probably the major cause of progressive neural damage in older children and adults with dysraphism lesions.
Clinical Manifestations
Bony spina bifida occulta at L5-S1 is a common radiologic finding in children and adults and is usually associated with no symptoms or signs. Unless other findings are present, no further evaluation or treatment is required. Signs of clinically significant spina bifida occulta may be in the form of cutaneous, neurologic, orthopaedic, urologic, or rectal abnormalities (Table 31–2).
TABLE 31–2 Presenting Symptoms and Signs of Occult Spinal Dysraphism
| % | |
|---|---|
| Leg weakness | 48 |
| Cutaneous abnormality | 48 |
| Foot deformity | 39 |
| Urinary incontinence | 36 |
| Fecal incontinence | 32 |
| Sensory abnormality | 32 |
| Recurrent urinary tract infection | 20 |
| Gait abnormality | 16 |
| Scoliosis | 14 |
Adapted from James H, Walsh J: Spinal dysraphism. Curr Prob Pediatr 11:1-25, 1981.
Cutaneous Syndrome
Cutaneous abnormalities indicative of an underlying spinal dysraphism are situated on or near the midline, usually in the lumbrosacral region, but occasionally in the thoracic region or the neck.66 A wide variety of abnormalities are seen.
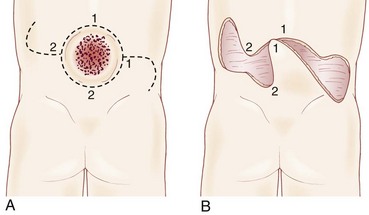
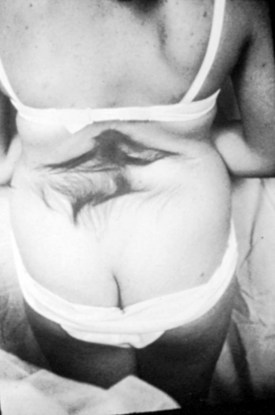
A striking finding is the hairy patch (hypertrichosis), also known as “faun’s tail,” which always occurs in the midline. It may be small but more commonly is a wide, diamond-shaped patch in the lumbar or lower thoracic area (Fig. 31–6). Frequently, the patient’s mother has trimmed the hair for years before presentation to the physician. If the dysraphism is in the cervical or upper thoracic region, there is usually a smaller patch of silky hair. The underlying abnormality may not be confined to the level of the hairy patch, and it is important to image the entire spine. Hypertrichosis may occur with any of the types of dysraphism but is particularly associated with split cord malformations.67
Subcutaneous lipomas at or near the midline of the lumbosacral spine may indicate an underlying intradural lipoma. These are nontender, poorly circumscribed soft masses of fat that are continuous with the normal subcutaneous tissue and with the intraspinal portion of a lipomyelomeningocele. The overlying skin is normal, hairy, or dimpled, and there may be an associated angioma or skin tag (Fig. 31–7). Other conditions may manifest as skin-covered masses overlying the caudal spine (Table 31–3). Often, an older patient gives a history of having had the superficial mass removed for cosmetic reasons.
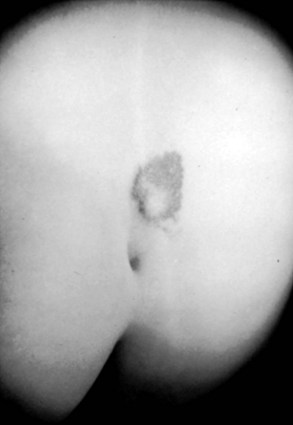
TABLE 31–3 Differential Diagnosis of Skin-Covered Lesions Overlying the Spine
Atretic meningoceles may also be seen, consisting of a central area of thin, pearly skin surrounded by a halo of red, pink, or brown (spinal aplasia cutis). These represent myelomeningoceles or meningoceles that have partially healed spontaneously. Dimples at the tip of the coccyx (coccygeal pits) are frequent findings in normal newborns and usually have no significance.68 Deep dimplelike depressions higher in the spine may be the external stigma of an epithelialized tract that connects with the filum terminale, spinal cord, or intraspinal dermoid cyst and require further investigation. The meningocele manqué is a skin blemish sometimes referred to as a “cigarette burn” because it resembles a small scar with loss of skin in the midline. When this blemish is obvious at birth, it may cause concern of CSF leakage, but this seldom occurs. Other cutaneous lesions that may be associated with a tethered cord include rudimentary caudal appendages (“tails”) and asymmetrical gluteal folds.
Neurologic Syndrome
Muscle weakness and gait disturbance are usually apparent at 2 years of age when a child begins to walk. On examination, there may be striking muscular atrophy and leg-length asymmetry. The deep tendon reflexes may be normal, increased, or absent, giving the pattern of a mixed upper and lower motor neuron lesion. Patchy sensory loss is present, particularly in the distal leg and perineum. Pain in the back with a radicular component is frequent in older children or adults.69
Orthopaedic Syndrome
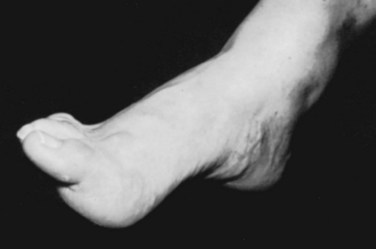
The most frequent orthopaedic finding is unilateral or bilateral cavovarus deformity of the foot, with or without leg-length discrepancy (Fig. 31–8). There may also be “claw toes.” The abnormal gait is the result of orthopaedic deformity and muscular weakness. It is presumed that the foot deformity is due to lack of, or weak innervation of, antagonistic muscles of the lower extremity.70 Scoliosis, especially if found in a young child or a boy or if noted to be rapidly progressive or accompanied by pain, also suggests an underlying spinal cause.
Urologic Syndrome
Occult spinal dysraphism should always be considered when one encounters infants with an abnormal voiding pattern, new-onset urinary or fecal incontinence in a previously toilet-trained child, or urinary tract infection in a child of any age. As opposed to older children, most infants with occult spinal dysraphism have normal results on urodynamic testing.71 This fact underscores the importance of recognizing the syndrome as early as possible so that prophylactic surgery can be performed.
Anorectal Anomalies
Anorectal anomalies, including vesicointestinal fissures, cloacal extrophy, rectovesical fistulas, and imperforate anus, are frequently associated with an underlying tethered cord.72 The overall incidence of tethered cord with imperforate anus is 35% and may be increased in boys.73 In many cases, no cutaneous abnormality is seen, and radiographic screening is indicated in these patients.
Radiologic Investigation
Spine MRI is the imaging study of choice to screen for occult spinal dysraphism. Bony anatomy is poorly seen but is of secondary importance in planning an operative approach for most lesions. A fibrous tract extending from a cutaneous coccygeal pit to the bony coccyx represents a persistent coccygeal ligament and is of no clinical significance. The presence of fat in the filum is a frequent incidental finding on MRI. Generally, if the conus is at the normal level (at or above the L2-3 interspace) and if the patient has no clinical signs or symptoms of a tethered cord, surgery is not warranted. Fat occurring very close to the conus may represent a special case, however, because this finding is associated with clinical tethering.74 A tethered cord may be suspected when the conus lies at the L2-3 level or below in a child or adult or if the conus is dorsally displaced.
Diffusion-weighted imaging may differentiate epidermoid cysts from arachnoid cysts.75 Postoperative studies may be difficult to interpret. The conus rarely ascends significantly after successful surgery, and even in asymptomatic patients the conus is shown to be dorsally displaced postoperatively. Although MRI is currently the best screening test for spinal cord tethering, it may not be useful for ruling out postoperative retethering. Some authors have proposed that imaging in the prone position to evaluate cord motion might be useful, but this has not been of value clinically.76,77
Ultrasonography through the infant spine or through congenital spina bifida or laminectomy defects has also been used to screen for occult spinal dysraphism.76,77 Although the level of the conus is readily seen, it is difficult to assign a specific spinal level to its termination. Sometimes one can visualize the tethering element, such as a hypertrophied filum or lipoma. Spinal cord motion has also been used to assess tethering.78 Ultrasonography may be useful as a screening test when the probability of a tethered cord is low, but MRI is preferable if an occult dysraphic lesion is strongly suspected.
Cases of tethered spinal cord with the conus in normal position on imaging studies have been described.79 Tethering is suspected on the basis of the typical clinical syndrome and confirmed at surgery. Mechanisms included hypertrophied filum terminale, meningocele manqué, and split cord malformation. This situation is uncommon, but when typical symptoms are present, surgical exploration is justified.
Specific Entities
Lipomyelomeningocele
Lipomyelomeningocele occurs at a rate of 1 in 4000 live births and in contemporary series is the most common cause of tethered cord syndrome.80 The term lipomyelomeningocele is misleading, in that it suggests herniation of neural elements through a spina bifida defect into a meningeal sac. The neural elements remain within the spinal canal, and only the lipoma itself may protrude through the spina bifida defect to manifest as a subcutaneous mass. Some authors have used the term lipoma of the cauda equina, but the fatty mass is invariably attached to the conus medullaris or filum terminale rather than to the cauda equina. Chapman81 classified lipomyelomeningoceles as lesions that attach to the caudal end of the conus, lesions that insert dorsally, and lesions that are transitional.
If the lipoma inserts onto the dorsal surface of the conus, there is usually a substantial subcutaneous mass, which may be asymmetrical. Along the lateral interface of the attachment of the lipoma to the spinal cord, the dura and pia are also fused (Fig. 31–9). Sensory roots emerge just anterior to this lateral line of fusion. As a result, neither the sensory roots nor the motor roots are actually within the lipoma. Alternatively, the lipoma may join the conus at its caudal end. The remaining mass may lie entirely within the spinal canal or extend dorsally through the spina bifida defect (Fig. 31–10). The fatty tumor may replace the filum terminale, or there may be a separate filum that lies anteriorly. The nerve roots usually lie ventral to the fatty mass but may lie within the fibrous ventral portion of the mass itself. Transitional forms may occur, which are often the most difficult to manage surgically. Probably the rarest subtype is the chaotic lipomyelomeningocele, described by Pang and colleagues,82 which has a prominent ventral component. MRI often defines the type of lipomyelomeningocele, which is helpful in understanding the anatomy that is encountered at operation.
< div class='tao-gold-member'>






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


