Fig. 2.1
Chilblain lupus erythematosus. (a) Symmetrically distributed, violet lesions with single erythematous papules and plaques involving acral locations of fingers. (b) Circumscribed, papulonodular, painful, bluish-red lesions at distal part of toes
Differential Diagnosis
The differential diagnoses include true cold-induced perniones or chilblains as well as cryoglobulinemia [5], which are often difficult to distinguish from chilblain lupus erythematosus. In addition, erythema nodosum, lupus pernio associated with sarcoidosis, or embolic events, as well as acral vasculitis/vasculopathy may also be considered [9, 10].
Histology
While the picture will often be diagnostic in typical cases, histology may secure the diagnosis in case of doubt. Histological findings of lesional skin in chilblain lupus erythematosus consist of vacuolar degeneration of the dermoepidermal junction with occasional single-cell necrosis and broadening of the basement membrane. In addition, a superficial and deep lymphocytic and histiocytic infiltration with periadnexial and perivascular distribution along with interface dermatitis is observed. Throughout the stratum reticulare, increased mucin deposits may occur. On direct immunofluorescence, granular deposits of immunoglobulins as well as complement within the basement membrane zone can be visualized [2, 11].
Molecular Genetics of Familial Chilblain Lupus
Unlike sporadic forms of the disease, familial chilblain lupus is inherited as an autosomal dominant trait manifesting in early childhood [2]. It is caused by heterozygous mutations of the TREX1 gene encoding three prime repair exonuclease [3, 4]. Thus, familial chilblain lupus represents the first monogenic form of cutaneous lupus erythematosus (CLE). Moreover, rare variants in the TREX1 gene are also found in patients with multifactorial systemic lupus erythematosus (SLE) [12], implicating TREX1-associated defects in nucleic acid metabolism in the pathogenesis of systemic autoimmunity.
TREX1, three prime repair exonuclease 1, is a ubiquitously expressed intracellular DNase with high specificity for single-stranded DNA (ssDNA) [13].
TREX1 mutations are associated with overlapping, but distinct inflammatory phenotypes underscoring the role of nucleic acid metabolism in immune regulation. Biallelic TREX1 mutations cause autosomal recessive Aicardi-Goutières syndrome, an early-onset encephalopathy resembling congenital viral infection, which is characterized by intracranial calcification and elevated interferon-α (IFNα) in cerebrospinal fluid [14]. Remarkably, some children with Aicardi-Goutières syndrome develop lupus-like symptoms over time [15]. Heterozygous TREX1 mutations have also been described in patients with autosomal dominant retinal vasculopathy with cerebral leukodystrophy, an adult-onset disorder characterized by central nervous system degeneration, retinal vasculopathy, and nephropathy [15], and in patients with multifactorial SLE. Thus, rare variants of the TREX1 gene confer a high risk for developing SLE [12].
TREX1 has been implicated in a number of biological processes that involve degradation of intracellular DNA. Loss of TREX1 function has been shown to impair DNA degradation during granzyme A-mediated apoptosis [3]. Apoptosis is highly relevant in the pathogenesis of different forms of lupus erythematosus. First, defective apoptosis may lead to impaired antigen-mediated B cell maturation and selection of autoreactive T cells, causing loss of self-tolerance. Secondly, self-antigens displayed on cell surfaces as a result of deficient disposal of extracellular nuclear waste can induce an autoimmune response. Finally, uncontrolled apoptosis is an effector mechanism responsible for autoimmune destruction of healthy cells. Moreover, recent evidence suggests that intracellular accumulation of ssDNA in TREX1-deficient cells is also caused by defective degradation of nucleic acids derived from chronic cell cycle checkpoint activation or from endogenous retroelements [16, 17]. Retroelements including endogenous retroviruses and L1 elements account for almost half of the human genome. They replicate through retrotransposition, a copy and paste mechanism which involves reverse transcription of an RNA intermediate [18]. Interestingly, TREX1 has been shown to degrade retroelement-derived ssDNA and to prevent retrotransposition of endogenous retroelements [16].
Nucleic acids exposed in a cell by microbial infection or during tissue damage can evoke immune responses. This is accomplished by membrane-associated receptors such as Toll-like receptors and a growing number of cytosolic sensors, such as RIG-1, DAI, and AIM2, which mediate recognition of danger-associated molecular patterns and initiate innate immune responses mainly through induction of IFNα [19]. These nucleic acid-sensing systems have evolved as a first line of defense against pathogens such as bacteria and viruses. Defects in nucleic acid metabolism may lead to an inappropriate activation of nucleic acid sensors. The ensuing induction of the innate immune response may, through its stimulatory effect on the adaptive immune system, eventually result in the phenotypic expression of an autoimmune disease. In fact, nucleic acids are among the key targets of the autoimmune attack in lupus erythematosus. Moreover, the autoimmune phenotype of TREX1 deficient mice can be rescued by intercrossing with mice deficient for interferon regulatory factor 3 (IRF3) or the IFNα receptor 1 (IFNaR1), indicating that the phenotype is type 1 IFN-dependent [16]. In view of the important role of IFNα pathways for the pathogenesis of lupus erythematosus [20], the TREX1-associated phenotypic spectrum paradigmatically highlights the interplay between the innate and the adaptive immune systems in the pathogenesis of systemic autoimmunity (Fig. 2.2).
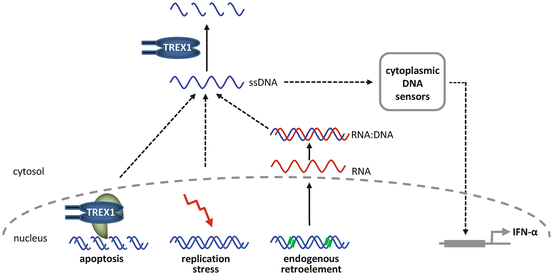
Fig. 2.2




Functional properties of TREX1 in nucleic acid metabolism. Nucleic acid species such as ssDNA generated during granzyme A-mediated apoptosis, DNA replication stress, or endogenous retroelement propagation are degraded by TREX1. Defects in TREX1 lead to accumulation of ssDNA and activation of nucleic acid sensors, which trigger an IFNα-mediated innate immune response
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
