93 Behçet’s Disease
Cutaneous lesions should display a neutrophilic vascular reaction on histopathologic examination.
Prognosis is variable, and patients typically have periods of exacerbations and remissions.
Behçet’s disease is a chronic, complex multisystem disease characterized clinically by oral aphthae, genital aphthae, cutaneous lesions, and ophthalmic, neurologic, or rheumatologic manifestations. The first description of Behçet’s disease was probably by Hippocrates in the fifth century bc,1 and the first modern account was presented in 1937 by the Turkish dermatologist Hulusi Behçet, who reported on a patient with recurrent oral and genital aphthae and uveitis.2
Epidemiology
Behçet’s disease is seen worldwide, with the highest prevalence reported in Turkey (80 to 370 patients per 100,000 inhabitants)3 and Japan (13.6 per 100,000).4 The prevalence and often the severity is increased in the Middle East and the Mediterranean (i.e., the “Silk Route”).5,6 The remainder of the Asian continent has a prevalence of 7 to 30 patients per 100,000 inhabitants.7 It is relatively uncommon in northern Europe (0.27 to 1.18 per 100,000 inhabitants) and the United States (0.12 to 0.33 patients per 100,000 inhabitants).3,5,7 Patients commonly fulfill the diagnostic criteria in their mid-20s to 30s.8 In the past, Behçet’s disease was thought to predominantly affect males, but current epidemiologic data show a more equal male-to-female ratio.9 Overall, the male-to-female ratio has decreased over the past 20 years with males being more affected in the Middle East and a female predominance existing in Korea, China, the United States, and northern Europe.7
Cause and Pathogenesis
Genetics
The onset of Behçet’s disease is believed to be sporadic, though familial clustering, families with multiple affected members, has been reported. Individuals with a first-degree relative with the disease are at an increased risk of developing the disease.10 Additionally, children of individuals with Behçet’s disease may have an earlier age of onset, suggesting genetic anticipation, which is due to a progressive increase in nucleotide repeats through consecutive generations.11 Familial occurrence differs regionally throughout the world and is more common in Korea, Israel, Turkey, and Arab countries, compared with Japan, China, and Europe.7
Studies have shown a significant association between the human leukocyte antigen (HLA)-B51 and Behçet’s disease.12,13 HLA-B51–positive patients are at an increased risk of developing Behçet’s disease (odds ratio of 5.9).14 This association is more common in the Middle East, Mediterranean, and Japan, though not seen as often in Western nations. Disease prognosis also appears to be more severe in HLA-B51–positive patients.7 The role of HLA-B51 in Behçet’s remains unclear. It may be that HLA-B51 is not directly involved in causing the disease but is closely linked to disease-related genes.15 Candidate genes have been localized to chromosome 6 and include the major histocompatibility complex class I chain-related gene A (MICA) and, more specifically, the MICA6 allele; perth block (PERB); new organization associated with HLA-B (NOB); and transporter associated with antigen processing genes (TAP).15,16 Other hypotheses suggest that HLA-B51 may contribute to the onset of Behçet’s disease by serving as a heterologous antigen either through original antigen presentation or through viral or bacterial molecular mimicry.15,17 A recent genome-wide association study confirmed the HLA-B51 relationship and also identified a second, independent association within the MHC class I region.18
Although Behçet’s disease has many features in common with the spondyloarthropathies, especially those associated with inflammatory bowel disease (IBD), the disorder in IBD patients generally evolves in a pattern resembling reactive arthritis, with an erosive axial arthritis; erosive arthritis and HLA-B27 are not associated with Behçet’s disease.19
Immune Mechanisms
Immune mechanisms play a major role in Behçet’s disease. Heat shock proteins, cytokines, alterations in neutrophil and macrophage activity, and autoimmune mechanisms have all been implicated.15 Heat shock proteins are released in response to stress and may be involved in stimulating a T helper type 1 immune response through interaction with Toll-like receptors.20 Specifically, immunoglobulin M–type, 47-kD cell surface heat shock protein against α-enolase has been identified in patients with Behçet’s disease.21 Although most of the T lymphocytes thought to be involved in this reaction are of the γδ type, the diversity of T lymphocytes seen in the disease suggests a response to multiple antigens, which may account for the various symptoms seen in Behçet’s.22 Cytokines such as interleukin (IL)-1, IL-8, IL-12, IL-17, and tumor necrosis factor (TNF) seem to be involved in the pathogenesis. Although elevated cytokine levels may serve as an indicator of disease severity, it should be appreciated that plasma TNF levels may rise and fall as an acute-phase reactant along with C-reactive protein and the erythrocyte sedimentation rate.23 The production of these proinflammatory cytokines, which are responsible for the chronic inflammation observed, may be the result of activated macrophages.15,24 In addition to macrophage activation, neutrophil chemotaxis and phagocytosis are increased in the lesions of Behçet’s disease.15,25 This increased activity of neutrophils leads to tissue injury in the form of the neutrophilic vascular reaction seen in lesions such as aphthae, pustular cutaneous lesions, and erythema nodosum–like lesions. Circulating immune complexes also play a role in precipitating the characteristic neutrophilic vascular reaction.26 Finally, the role of endothelial cell dysfunction in the pathogenesis of Behçet’s disease has been suggested by decreased levels of prostacyclin in the serum of Behçet’s disease patients. Increased nitric oxide concentrations in the serum, synovial fluid, and aqueous humor of individuals with Behçet’s may play a role in endothelial activation, resulting in vascular inflammation and thrombosis.27,28 Elevated homocysteine levels have been cited as the cause of the increased nitric oxide concentrations, suggesting that hyperhomocysteinemia may represent an acquired risk factor for Behçet’s, which is potentially reversible.29–31
Infectious Agents
Several studies have suggested a role for various infectious agents in the pathogenesis of Behçet’s disease; however, no organisms have been consistently isolated. Antistreptococcal antibodies have been isolated in the serum of patients with Behçet’s disease.32 Higher concentrations of Streptococcus sanguis have also been found in the oral flora of patients with Behçet’s disease and may play a role in the development of aphthae, which is often the initial manifestation.33 In addition to streptococcal antigens, other bacteria such as Escherichia coli and Staphylococcus aureus may have a role in Behçet’s disease through the activation of lymphocytes.34 Additionally, a lipoprotein of Mycoplasma fermentans has been found in patients with Behçet’s. This lipoprotein (MALP-404) contains the specific peptide motif, which is capable of being presented by HLA-B51.35 Studies have also indicated that there is a higher rate of Helicobacter pylori cytotoxin-associated gene-A antibodies in Behçet’s patients. These antibodies may cause endothelial damage via cross-reaction with endothelial antigens. H. pylori eradication in these patients has been shown to decrease disease severity.36
Herpes simplex virus (HSV) deoxyribonucleic acid (DNA) has been isolated from the nuclei of peripheral blood lymphocytes by polymerase chain reaction (PCR) assay in patients with Behçet’s disease.19 HSV has also been detected by PCR in biopsy samples of genital and intestinal ulcers of Behçet’s patients.34 Other studies, however, have shown no difference in the detection of HSV in Behçet’s patients with and without oral aphthae.37
In summary, although the cause and pathogenesis of Behçet’s disease are not completely understood, they likely involve an infectious or environmental trigger and subsequent inflammatory response in a genetically predisposed individual. The article by Zouboulis and May15 provides an excellent overview of the current understanding of the pathogenesis of Behçet’s disease.
Clinical Features
Aphthae
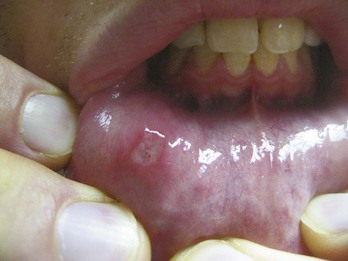
Oral aphthae, or canker sores (Figure 93-1), are often the initial feature of Behçet’s disease and constitute a requisite diagnostic feature (although many believe that Behçet’s occurs in the absence of oral aphthae). Oral ulcerations usually occur in crops of more than 3 to 10s of lesions, but individual lesions may occur on the buccal mucosa, gingiva, lips, and tongue. Aphthae tend to be painful and shallow, and they heal without scarring over 1 to 3 weeks.38 Genital ulcers typically occur on the scrotum and penis in males and on the vulva or vaginal mucosa in females. These aphthae are similar in appearance to oral lesions, but they have a greater tendency to scar and may recur less frequently.38 Lesions in the oral mucosa are generally easy to distinguish from oral HSV, but with genital lesions, HSV should be excluded by viral culture or PCR before lesions are accepted as a diagnostic criterion.
Cutaneous Lesions
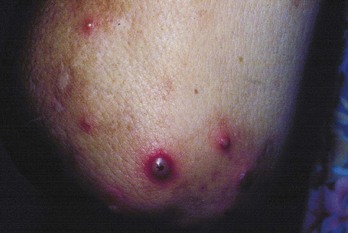
Several cutaneous manifestations of Behçet’s disease have been described: erythema nodosum–like lesions, pyoderma gangrenosum–like lesions, Sweet’s syndrome–like lesions, cutaneous small vessel vasculitis, and pustular vasculitic lesions (Figure 93-2) including lesions induced by trauma—the so-called pathergy lesion.38 Pathergy signifies the development of erythematous pustules or papules 24 to 48 hours following puncture of the skin with a 20- to 21-gauge sterile needle.39 Specimens from all these lesions demonstrate a neutrophilic vascular reaction on histopathologic analysis.40 Acneiform or pseudofolliculitis lesions should be considered nonspecific, nondiagnostic findings because of their common occurrence in acne vulgaris and folliculitis.
Ophthalmic Features
A variety of ocular manifestations have been reported in Behçet’s patients including anterior and posterior uveitis, retinal vasculitis, and hypopyon, with secondary glaucoma, cataract formation, decreased visual acuity, and synechiae formation.41 Ocular involvement occurs in 83% to 95% of men and 67% to 73% of women with Behçet’s disease.41 Although ocular involvement is not commonly the presenting feature of Behçet’s disease, it is a major source of serious morbidity, and close ophthalmologic evaluation and follow-up are critical to prevent blindness in these patients.42 BenEzra and Cohen43 suggested that if ocular disease does not present within a few years of diagnosis, it is unlikely to be a major problem.
Arthritis
The arthritis of Behçet’s disease is typically a nonerosive, inflammatory, symmetric, or asymmetric oligoarthritis, although polyarticular and monoarticular forms are also seen. The most commonly involved joints are the knees, wrists, ankles, and elbows.44 The prevalence of arthritis among different populations ranges from 40% to 60%, and joint erosions are not observed.42 Dilsen and colleagues45 reported that 10% of patients with Behçet’s disease had a sacroiliitis. However, HLA-B27–positive patients were not excluded from their series, and occult IBD was not excluded, as required by O’Duffy and Goldstein.46 Other studies have shown no significant difference in the occurrence of sacroiliitis between patients with Behçet’s disease and the normal population. HLA-B27–positive patients with erosive sacroiliitis should be included in the reactive arthritis or enteropathic arthritis disease spectrum, given the erosive, axial nature of the arthritis in the HLA-B27 pattern. This contrasts with the classically nonerosive, nonaxial nature of the arthritis in Behçet’s disease. Oral aphthae, ocular lesions, erythema nodosum–like lesions, pustular vasculitis, and pyoderma gangrenosum all occur in patients with IBD.
Other Systemic Manifestations
Central nervous system (CNS) involvement is most commonly characterized by brain stem or corticospinal tract syndromes (neuro-Behçet’s syndrome), venous sinus thrombosis, increased intracranial pressure secondary to venous sinus thrombosis or aseptic meningitis, isolated behavioral symptoms, or isolated headache.47 Rarely, ruptured aneurysms, peripheral neuropathy, optic neuritis, and vestibular involvement can occur.47 Poor prognosis is associated with a progressive course, parenchymal or brain stem involvement, cerebellar symptoms, and cerebrospinal fluid abnormalities.48 Cranial and peripheral nerve involvement may also occur.
Patients with Behçet’s disease may have gastrointestinal lesions resembling orogenital aphthae. These occur most commonly in the ileocecal region and in the ascending colon, transverse colon, or esophagus.30 Large aphthae may lead to perforation. Presenting symptoms include abdominal pain, diarrhea, and melena. It is important to distinguish IBD from Behçet’s disease.49 Aphthae may also affect the bladder.
Pulmonary abnormalities are uncommon in Behçet’s disease. Pulmonary artery aneurysms occur most frequently, followed by other complications secondary to vasculitis affecting the small pulmonary vessels. Aneurysm, thrombosis, hemorrhage, and infarction can result and cause death in patients with Behçet’s disease.31
Renal manifestations are not common. They vary from minimal changes to proliferative glomerulonephritis and rapidly progressive crescentic glomerulonephritis. The pathogenesis likely involves immune complex deposition.50
Cardiac complications include myocardial infarction, pericarditis, arterial and venous thromboses, and aneurysm formation. Thromboses more commonly involve the venous system, sometimes leading to superior and inferior vena cava obstruction.40 Cardiac manifestations, either occlusive or aneurysmal, are postulated to occur due to a vasculitis of the vasa vasorum, which induces a thickening of the media and splitting of elastic fibers.51 Atherosclerosis does not appear to occur at an increased rate, as is seen in many autoimmune diseases such as systemic lupus erythematosus.52 Mortality in Behçet’s disease is low and is usually related to pulmonary or CNS involvement or to bowel perforation.9
Limited data are available evaluating Behçet’s in pregnancy. One case-control study reported more remissions than exacerbations during and after pregnancy with higher rates of pregnancy complications but no changes in neonatal outcomes.53
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


