CHAPTER 75 Basic Science of Spinal Cord Injury
Pathophysiologic Response to Spinal Cord Injury
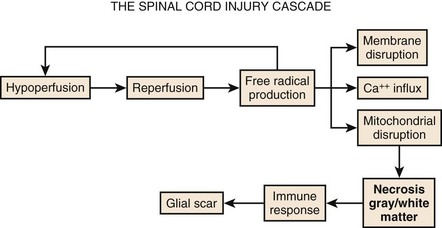
Although physical trauma to the spinal cord undoubtedly causes direct physical injury to the neural tissues, it has become clear that a cascade of events follows the inciting episode and can both inhibit recovery and cause additional neural damage.1,2 This concept of secondary injury has been validated in both animal and clinical studies, and secondary injury mechanisms have been the target of the bulk of pharmacologic interventions to date. These processes begin immediately following the injury and continue for weeks (Fig. 75–1).
Circulatory Collapse
The cascade of secondary injury often begins with microcirculatory insufficiency shortly after mechanical trauma.2 Vascular hypoperfusion due to capillary loss, capillary spasm, thrombosis, systemic hypotension, and autonomic regulatory interruption lead to cellular ischemia at the epicenter of cord injury. The gray matter, rich with neuronal cell bodies, is highly vulnerable to ischemia. The resultant shift in pH renders neuronal cell body and axonal membranes highly vulnerable to subsequent injury. The neurons are now primed to enter into what will become an increasingly self-destructive path.
Oxidative Damage
After a transient period of hypoperfusion and ischemia, a sudden and unregulated reperfusion of the injury epicenter occurs. The introduction of oxygen to the compromised cell membranes produces a highly toxic environment whereby the membrane lipid fatty acids undergo oxidation.3 This membrane lipid peroxidation produces several varieties of free radicals that in turn drive even further lipid peroxidation and free radical production. Some of the free radicals accumulate within the cell and denature deoxyribonucleic acid (DNA) and mitochondrial proteins, and eventually bring energy production to a halt resulting in irreversible damage and cell death.
Excitotoxicity
The remainder of the free radicals further disrupt and destabilize the neuronal membrane. The cellular release of the ubiquitous neurotransmitter glutamate changes the extracellular space into a hostile extracellular milieu.4 Glutamate activates various cell surface receptors that in turn mediate a large variety of intracellular processes. Excessive glutamate will drive these processes to the point of fatal overload to the cell. The most studied of the glutamic receptors is the N-methyl-D-aspartate (NMDA) receptor, which mediates entry of Ca++ into cellular cytoplasm from both extracellular and intracellular stores. Although calcium in physiologic amounts is the necessary component for many important enzyme-mediated cellular processes, pathophysiologic quantities of calcium lead to the persistent activity of destructive enzymes including lipoxygenases and phospholipases. These enzymes will again target the beleaguered cell membrane to generate free radicals from lipid oxidation. The radicals will disrupt cellular proteins and, in particular, those that mediate the ability of mitochondria to drive oxidative phosphorylation. The neuron, starved of ATP, will terminate itself via necrotic or apoptotic cell death mechanisms.
Neuroimmunologic Response
The events taking place moments after spinal trauma, beginning with microcirculatory failure within the cord and leading to free-radical-mediated cytotoxicity of the gray and white matter, do not go unnoticed by the immune system. The ever-vigilant inflammatory cells of the body are instantly attracted to the neuronal self-destruction. Over the next hours to weeks, they will lay the foundations for an extracellular environment that will inhibit axonal regeneration. The first of these cells to appear at the site of injury are circulating neutrophils. Once active, neutrophils will secrete cytokines that stimulate production of phospholipases and cyclooxygenase.5 The former will consume neural membranes to produce arachidonic acid, which the latter (cyclooxygenase) uses to produce prostaglandins and thromboxanes. Prostaglandins (PGE2, PGD2, PGF2a, PGI2) serve (1) to amplify the inflammatory response by increasing capillary permeability to allow additional inflammatory cells influx; (2) to increase neuronal calcium concentration, thus promoting excitotoxicity; and (3) to activate other inflammatory cells.1 Thromboxanes promote platelet aggregation within capillaries and thus worsen local tissue ischemia.
Astroglial Scar
Trauma to the adult spinal cord is particularly devastating because of the inability of the central nervous system (CNS) to regenerate after injury. Unlike in the peripheral nervous system, axonal recovery in the spinal cord is thwarted by two fundamental obstacles: the inherently weak regenerative ability of CNS axons and a powerfully inhibitive postinjury milieu of physical and chemical factors.6 The most potent of these factors is the glial scar that develops after any CNS injury.7–9 The glial scar is a collection of reactive cells (astrocytes, microglia, oligodendrocyte precursors, meningeal fibroblasts) that express cell-surface and matrix molecules, which surround the area of injury and ultimately repel the advancement of regenerating axons (Fig. 75–2).
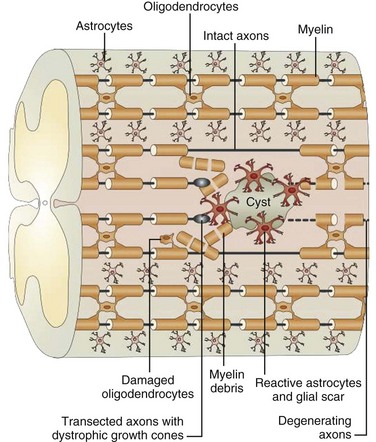
The scar features a core zone of meningeal cells and oligodendrocyte precursors, as well as a peripheral zone of astrocytes, oligodendrocyte precursors, and microglia. The core zone is separated from the surround zone by a basement membrane composed mostly of type IV collagen.10 Although some axons may regenerate through the surround zone, no axon can penetrate the core zone without some form of experimental manipulation.11
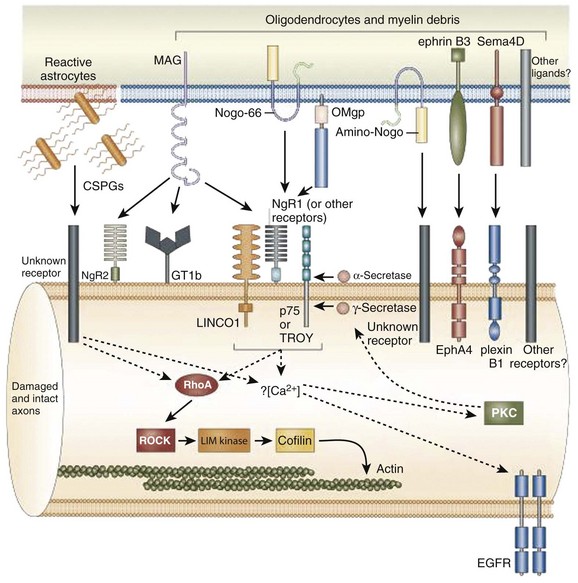
The inhibitory effects of the scar are conferred by three classes of molecules, all of which are expressed by one or more of the reactive cells in the glial scar. These include the chondroitin sulphate proteoglycans (CSPGs) (NG2, brevican, phosphacan, neurocan, versican), semaphorin 3 proteins, and eph/ephrin tyrosine kinases. Although the precise mechanisms of their actions are unclear, the molecules exert their inhibitory effects either by directly or indirectly binding to the axon cell surface or by binding and deactivating trophic factors, cell adhesion molecules, and extracellular matrix molecules that are a requisite for axonal growth and regeneration.12 The ultimate effect of the gliotic response to injury is the inhibition of successful axonal regeneration and remyelination by both physical and chemical means (Fig. 75–3).13
There is tremendous therapeutic potential in the ability to modulate the gliotic scarring response to CNS injury. In-vitro and in-vivo studies to date, though relatively limited, have demonstrated enhancement of axonal regeneration and functional recovery after inhibition of specific glial scar constituents. Enzymatic digestion of the glycosaminoglycan chains of CSPGs, for instance, stimulates axonal regeneration through the site of injury.14 Function-blocking antibodies to semaphorin receptors have allowed sensory axons to regenerate into the formidable core zone of the scar.15 Chelating agents that prevent collagen IV synthesis around the core zone have also allowed successful axonal regeneration in some animal models.16 Animals with clonal deletions of a certain eph molecule have almost no astroglial scar response and demonstrate unimpeded regeneration of motor axons through the zone of injury.17
Despite its multifaceted inhibitory influence, several recent studies suggest that the glial scar must offer some protective benefit to the injured spinal cord. The role of the gliotic response in mitigating the extension of cord injury beyond the initial site of trauma, for instance, has been proposed.18 In a study of transgenic mice, selective ablation of reactive astrocytes in the glial scar after both contusion and penetrating SCI led to markedly increased tissue disruption, cellular degeneration, cystic changes, and profound and persistent motor deficits relative to nonablated controls.19 It is likely that both the cellular and extracellular matrix elements of the gliotic scar play a critical role in biochemical protection and structural stabilization of cord integrity, and thus function, after spinal cord injury.
Basic Science of a Cure
Despite the confounding nature of the injury response, several glimpses of hope for a cure exist and they capitalize on our understanding of the basic science of spinal cord injury. Consistent with their basic science foundations, the human acute SCI therapies have one of two ambitions: (1) to limit secondary injury (neuroprotection, acute surgical intervention, rehabilitation trials) and (2) to reverse injury (regeneration trials) (Box 75–1).
Methylprednisolone
The first randomized, controlled multicenter trial of a neuroprotective agent was the National Acute Spinal Cord Injury (NASCIS) I Study, which attempted to establish the clinical efficacy of methylprednisolone.20,21 Though its precise mechanism of action is still unclear, methylprednisolone was thought to exert either a cell-stabilizing effect via the glucocorticoid receptor or a cord-stabilizing effect via free-radical inhibition.22–24 The trial was based on several animals studies that suggested improved neurologic recovery when the corticosteroid was administered promptly after experimental injury.25 Published in 1984, NASCIS I included 330 patients with acute SCI (defined as any loss of sensation or motor function below the level of injury) who were randomized into two groups within 48 hours of injury: a “low-dose group” receiving 100 mg IV methylprednisolone bolus and then 25 mg every 6 hours for 10 days and a “high-dose” group receiving bolus and maintenance doses 10 times those of the low-dose group every 6 hours for 10 days. Outcome measures consisted of motor and sensory indices of 14 muscle groups and 29 dermatomes. The follow-up periods were 6 weeks and 6 months. Though there was no difference between the two groups in terms of neurologic outcome, there was an increased incidence of wound infection and even fatality in the higher-dose group, with the former achieving statistical significance.
One year later, NASCIS II was launched as the first randomized, prospective, placebo-controlled trial of a candidate therapy for SCI.26,27 It was devised to address the lack of a placebo control in NASCIS I and to incorporate new basic-science findings regarding effective methylprednisolone dosing and mechanisms of action. The study involved 487 patients who were randomized into three groups within 12 hours of sustaining either complete or incomplete SCI: a methylprednisolone group receiving an unprecedented 30-mg/kg IV bolus, followed by a maintenance infusion of 5.4 mg/kg/hr over 23 hours; a placebo group; and a third group consisting of a 5.4 mg/kg bolus and 4 mg/kg/hr maintenance infusion of the opioid antagonist naloxone, whose neuroprotective effects had also been suggested by animal studies.28 The outcome methodology was similar to that in NASCIS I.
When all members of the methylprednisolone group were compared with placebo, there were no statistically significant improvements in sensory or motor function at 6 weeks. However, when the steroid group was stratified according to timing of administration, patients receiving treatment within 8 hours of injury demonstrated a significant improvement in sensory and motor function by 6 weeks versus placebo. The relative improvement was sustained through the 6-month follow-up point. Within the steroid group treated within 8 hours, further subgroup analysis with respect to American Spinal Injury Association (ASIA) scale injury severity revealed that class A patients had the greatest statistically significant improvement in motor and sensory measures versus placebo. Additionally, ASIA C&D patients had only motor improvement, whereas class B patients had neither sensory or motor improvement that reached statistical significance.26,29 The complication of wound infection was more frequent but statistically insignificant in the steroid group despite the heavy dosing. There were no differences in motor or sensory outcomes between the naloxone and placebo group at either of the follow-up points. NASCIS II established the now ubiquitous “steroid protocol,” despite controversies regarding possible nontransparency, data misinterpretation, and near-normal function of some participants.
The objectives of the third and final NASCIS were to investigate the interplay between timing of steroid administration and duration of therapy and to evaluate the efficacy of the 21-aminosteroid tirilazad mesylate, which purportedly had a better safety profile than methylprednisolone. Four-hundred ninety-nine patients were randomized into three treatment groups within 6 hours of injury: the first group received methylprednisolone according to the NASCIS II dosing for 24 hours, the second group received this dosing for 48 hours, and the third group received a methylprednisolone bolus of 5.4 mg/kg/hr followed by a maintenance infusion of tirilazad at 2.5 mg/kg IV every 6 hours for 48 hours.30 With outcome measures including motor function, sensory function, and functional independence; the NASCIS III revealed that increased duration of steroid administration (48 hours) resulted in statistically significant benefit only if treatment was initiated between 3 and 8 hours of injury. Infectious complications were more common in the 48-hour corticosteroid group but were statistically insignificant. There were no differences between the tirilazad group and the 24-hour methylprednisolone group.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


