Chapter 21 Autoantigenesis and Antigen-Based Therapy and Vaccination in SLE
The discovery in the early 1990s that self-antigen–reactive T cells can be tolerized to prevent systemic lupus erythematosus (SLE) in animal models1 eventually led to clinical trials of autoantigenic peptides in patients with SLE.2–4 In fact, the U.S. Food and Drug Administration (FDA) has granted the “fast track” approval to start a phase 3 trial of Lupuzor, a posttranslationally modified peptide analog derived from U1-70K small nuclear ribonucleoprotein (snRNP).2 Although this development is encouraging, the identity of true autoantigen-reactive T cells and their exact role in SLE remains to be fully understood. Advances in use of in situ tetramer staining to identify human antigen–specific T cells in the affected organs of patients5 provide hope for identification of autoantigen-specific T cells that infiltrate the diseased organs in humans. Such T cells and their autoantigens can be true targets for therapy. In the meantime, growing evidence supports a role for apoptosis, posttranslational and other modifications in the antigens themselves, and determinant spreading as possible sources of autoantigens in SLE.6,7 Antigen mimicry has long been considered a mechanism of autoantigenesis. Molecular mimicry at the T-cell epitope level was detected between lupus-associated autoantigen Sm-D and microbial peptides.8 Exposure to naturally occurring hydrocarbon oils in otherwise normal mouse strains elicits chronic inflammation, which eventuates in a plethora of autoimmune manifestations, including nephritis, arthritis, pulmonary vasculitis, and lupuslike autoantibodies.9–12 Development of inflammation long before the onset of autoimmunity in these mice is believed to trigger the autoantigenicity of a variety of nuclear and cytoplasmic antigens.
Major autoantigens in SLE include DNA-associated antigens, namely, nucleosome, which is made up of double-stranded DNA (dsDNA) bound to the five histone molecules, H1, H2A, H2B, H3, and H4, and high-mobility group box 1 (HMGB1) protein, and RNA-associated antigens such as U1 small nuclear ribonucleoprotein (U1 snRNP), Ro/la complex, and ribosome.7 Studies suggesting a key role for dsDNA in eliciting inflammation in the pathogenesis of SLE have also rekindled interest in manipulating DNA structure using topoisomerase I inhibition, administration of DNase I, or modification of histones using heparin or histone deacetylase inhibitors as possible therapeutic options.13
Autoantigenesis: Mechanisms that Make an Antigen an Autoantigen
That only certain self-proteins frequently elicit an autoimmune response has intrigued many investigators to speculate that autoimmunity might occur as a result of altered self or modified self.14,15 In this section, we describe mechanisms—defective apoptosis, impaired removal of apoptotic cells, somatic mutations, genetic polymorphisms, alternative splicing, and posttranslational modifications—that could generate epitopes for which the immune system is not tolerized.16 See Box 21-1 for a summary of characteristics of autoantigens. The modified antigens can be taken up, processed, and presented by antigen-presenting cells (APCs) and recognized by existing potentially self-reactive B and T cells, resulting in breakage of tolerance and induction of autoimmunity. Defects in this pathway, that is, sensing and uptake and processing of antigens, can also lead to autoantigenesis. Finally, a bystander enrollment as an autoantigen can occur during epitope spreading of immune responses and as a result of mimicry with a foreign antigen.
Box 21-1
Some Common Features of Autoantigens Described in Lupus*
2. Evolutionarily conserved14,15
3. Genetically polymorphic24,162
4. Expressed in apoptotic blebs160
5. Restricted polyclonality, i.e., against autoantigens that are structurally or functionally related152
6. Restricted polyclonality through shared T-cell determinants among variable regions of different autoantibodies76,77
7. Posttranslationally modified29,168
8. Substrates of apoptotic enzymes (caspases)30,32
9. Mutated somatically22,23,157
10. Charged or coil-coil structure162
11. Molecular mimics of infectious agents33,34
Defective Apoptosis
Defective apoptosis can result in the generation of neoepitopes.13 Proteolytic cleavage of lupus-associated autoantigens, like poly (ADP-ribose) polymerase and a catalytic subunit of DNA-dependent protein kinase (DNA-PKCs), has been shown to disturb homeostasis and cause increased apoptosis. As a result of nuclear fragmentation and membrane blebbing in apoptosis, autoantigens that are targeted in SLE are reorganized and transported to cell surfaces.17 Secondary necrosis can also be an additional source of proteolytically modified forms of specific autoantigens.18 For example, during the initial apoptotic stages, several autoantigens, including poly ADP-ribose, are cleaved into apoptosis fragments. The apoptotic cells then undergo secondary necrosis in the absence of phagocytosis with additional modifications of autoantigens.18 A misguided immune response to these modified nuclear and cytoplasmic antigens is believed to be a major mechanism underlying autoantibody production in SLE.
Impaired Removal of Apoptotic Cells
Impaired removal of apoptotic cells could contribute to an overload of autoantigens (particularly nucleosomes) in circulation or in target tissues that could become available to initiate an autoimmune response. Nucleosomes are formed during apoptosis by organized cleavage of chromatin. These nucleosomes together with other autoantigens cluster in apoptotic bodies at the surfaces of apoptotic cells. Systemic release of these autoantigens is normally prevented by swift removal of apoptotic cells. However, if excessive apoptosis exceeds the rate of removal of apoptotic bodies, nucleosomes are released. A number of studies have identified abnormalities that lead to impairment of apoptotic debris removal in patients with SLE and in mouse models. These include deficiencies in complement components, particularly C1q, C2, and C419 as well as macrophage proteins that are pertinent to clearance of debris, including scavenger receptor A (SR-A), macrophage receptor and collagenous structure (MARCO),20 and mer tyrosine kinase.21
Mutations
Mutations in self-antigens, which may create a neoepitope, might trigger autoimmunity. For example, in a complementary DNA (cDNA) library made from peripheral blood lymphocytes (PBLs) of a patient with primary Sjögren syndrome, one study identified a deletion of an (A)-residue in a cDNA encoding for the nuclear autoantigen La (SSB). This leads to a frame shift mutation in one of the major autoepitope regions of the La antigen.22,23 The modified La peptide shared homology with (1) La protein itself and (2) a series of DNA-binding proteins, including other autoantigens, and viral proteins such as topoisomerase I, RNA-dependent RNA polymerase of influenza virus, and reverse transcriptase. The mutant La peptide represents a putative neoepitope that could be involved in triggering of the autoimmune response.
Genetic Polymorphisms
Genetic polymorphisms may create autoantigens.24 Analysis of sequence variability has revealed significantly more single-nucleotide polymorphisms (SNPs) within coding regions of known human autoantigens (n = 348) than of other human genes (n = 14,881). Autoantigens had 7.2 SNPs per gene, compared with 3.6 SNPs per control gene. As an example, human Ro52, a major autoantigen in rheumatic diseases, contains two synonymous and three nonsynonymous SNPs, and one of the nonsynonymous SNPs is located in the central immunodominant region of the autoantigen.25 Further, an intronic SNP that leads to aberrant splicing of Ro52 messenger RNA (mRNA), resulting in the generation of a shortened version of the Ro52 protein, is strongly associated with anti-Ro52 autoantibodies in primary Sjögren syndrome.26
Alternative Splicing
Alternative splicing can create autoantigens.16 A bioinformatics analysis revealed alternative splicing in 100% transcripts of 45 randomly selected autoantigens, which is significantly higher than the approximately 42% rate of alternative splicing observed in 9554 randomly selected human gene transcripts. Within the isoform-specific regions of the autoantigens, 92% and 88% encoded major histocompatibility complex (MHC) class I– and class II–restricted T-cell antigen epitopes, respectively, and 70% encoded antibody-binding domains. Furthermore, 80% of the autoantigen transcripts underwent noncanonical alternative splicing, a rate that is also significantly higher than the less than 1% rate observed in randomly selected gene transcripts.
Posttranslational Modifications
Posttranslational modifications in a protein could also act as a means to promote autoreactivity.6,7,15 Between 50% and 90% of the proteins in the human body acquire posttranslational modification. Many of these modifications are necessary for the biological functions of proteins. Some posttranslational modifications, however, can create new self-antigens by altering immunologic processing and presentation. Because these modifications occur after the lymphocyte has undergone negative selection, the existing B and T lymphocytes can recognize the modified antigens, thus causing tolerance breakdown. For example, the spontaneous conversion of asparagine or aspartic acid residues to isoaspartyl residue renders cytochrome c and snRNP D peptides immunogenic in murine models of SLE. Mice develop T-cell responses to the isoaspartic acid–containing peptides but not to the native aspartic acid–containing peptides. Autoantibodies in these mice, however, recognize both the isoaspartic peptides and the native aspartic acid peptides. Isoaspartic acid residues have also been detected in histone H2B, a common autoantigen in spontaneous and drug-induced lupus.27 In other examples of a likely role of posttranslational modification in autoimmunity, patients with SLE have been found to have autoantibodies against the C-terminus of snRNP that contains symmetrical dimethyl arginines,28 and phosphorylated serine/arginine–rich residues of the SR protein (a family of pre-mRNA splicing factors). Interestingly, some autoantibodies are directed at dephosphorylated SR proteins that normally would exist in a phosphorylated state.29
Altered Antigen Processing
Altered antigen processing can lead to generation of new autoantigens for which the immune system is not tolerized. In xenobiotic models of lupuslike autoimmunity, cell death following exposure to autoimmunity-inducing agents leads to generation of novel protein fragments that may activate self-reactive T lymphocytes.30 During apoptosis, interaction of several autoantigens with granzyme B has been shown to generate unique protein fragments that are not observed during any other form of cell death. Interestingly, nonautoantigens are either not cleaved by granzyme B or are cleaved to generate fragments identical to those formed in other forms of apoptosis. Therefore the ability of granzyme B to generate unique fragments appears to be an exclusive property of autoantigens.31,32
Molecular Mimicry
Molecular mimicry has been proposed to explain the role of microbial antigens in inducing and/or exacerbating autoimmune diseases. Association between the development of SLE and viruses such as Epstein-Barr virus (EBV), Coxsackie virus, and retroviruses like human T-lymphocyte virus (HTLV) has been described.33 For example, analysis of autoantibody responses in patients with SLE prior to the onset of clinical disease led to identification of an initial autoantigenic epitope that appears in some patients who have antibodies to 60-kDa Ro antigen. This initial epitope cross-reacts with a peptide from the latent viral protein Epstein-Barr virus nuclear antigen 1 (EBNA-1). Animals immunized either with the initial epitope of 60-kDa Ro or with the cross-reactive EBNA-1 epitope progressively develop autoantibodies binding to multiple epitopes of Ro and spliceosomal autoantigens. The immunized animals eventually demonstrate clinical symptoms of lupus, thus providing a strong evidence for association of EBV infection and development of SLE.34 One study has demonstrated molecular mimicry at the T-cell epitope level between lupus-associated autoantigen Sm-D and microbial peptides (Table 21-1). The researchers found that distinct autoreactive T-cell clones were activated by different microbial peptides, suggesting a role for molecular mimicry at the T-cell epitope level for activation of autoantibody augmenting autoreactive T cells.8


Defective Sensing and Uptake of Autoantigen
Accumulating evidence suggests that under autoimmune conditions, DNA fragments alone are able to induce signaling cascades that promote inflammation. Pathways through which self-DNA is able to induce proinflammatory reactions are distinct from those activated by microbial nucleic acids.13 The dsDNA-containing immune complexes undergo endocytosis after engaging the B-cell receptor (BCR) on B cells or Fc receptors (FcRs) on dendritic cells (DCs), macrophages, and glomerular cells. Additionally, dsDNA can be internalized after binding to LL-37 (cathelicidin), or through the HMGB1-RAGE (receptor for advanced glycation end-products) pathway. These routes result in localization of DNA to endosomes. The dsDNA-sensing pathways activated differ according to the structure of the DNA. CpG-rich dsDNA activates Toll-like receptor 9 (TLR9), whereas AT-rich dsDNA signals through DAI (DNA-dependent activator of interferons) or RNA polymerase III. These signaling pathways all lead to production of type I interferon (IFN) and inflammation. A fourth dsDNA-sensing pathway involves the AIM2 inflammasome and results in activation of interleukin-1 beta (IL-1β) and induction of pyroptosis. It is logical to hypothesize that defects in these pathways may trigger autoantigenicity of DNA,13 although little is known about the exact role these pathways play in the pathogenesis of SLE.
Chronic Inflammation as a Trigger of Autoantigenesis
Exposure to naturally occurring hydrocarbon oils, such as the medium-length alkane 2,6,10,14-tetramethyl pentadecane (TMPD, also known as pristane), is associated with the development of chronic inflammation and a variety of pathologic findings in humans and animal models.9,35 Depending on the genetic background, persistent inflammation in otherwise normal strains of mice eventuates in a cascade of events leading to a plethora of autoimmune manifestations, including glomerulonephritis, arthritis, pulmonary vasculitis, and autoantibodies against a variety of nuclear and cytoplasmic antigens, which mimics human SLE syndrome more closely than the genetically susceptible strains of mice.9–11
Data suggest that different autoantibody subsets and organ injury are mediated through different pathways, and both innate and adaptive immune responses participate in the development of full lupuslike syndrome in TMPD-injected mice.12 The initial response to TMPD is orchestrated by major components of the innate immune system. It starts with the infiltration of neutrophils and Ly6Chi inflammatory monocytes into the peritoneal cavity, which lasts for several months.35 Type I IFN (IFN-I) production downstream of TLR7 signaling and CCR2 (chemokine [C-C motif] receptor 2) plays a role in the influx of monocytes, whereas IL-1α and CXCL5 (chemokine [C-X-C motif] ligand 5) play a role in neutrophil recruitment to the peritoneal cavity. The adaptor molecules MyD88, IL-1R–associated kinase 4 (IRAK-4), IRAK-1, and IRAK-2 play a role in the recruitment of both monocytes and neutrophils. Deficiency of IL-6, TLR9, and TLR4 attenuate organ injury and production of anti-dsDNA and/or anti-RNP autoantibodies. Although the exact cascade of events that lead to autoantigenicity of lupus autoantigens is not known, studies in this model suggest a role for chronic inflammation in autoantigenesis.
Mechanisms by which Autoantigens May Contribute to the Development of Disease
Induction of Effector T cells
Autoantigen-specific T cells can contribute to pathogenesis of SLE by helping B cells produce autoantibodies or by directly infiltrating the tissues. Ample evidence also supports the requirement of T-cell help for pathogenic autoantibody production.36,37 This help is provided by T cells that react with peptides derived from various autoantigens.37–43 One study has shown that MRL-lpr mice that have no secreted immunoglobulin (Ig) develop spontaneous T-cell activation and some disease,44 suggesting a direct, antibody-independent role for T cells in the development of SLE-like disease. “Autoantigen-specific” T cells, however, have not yet been demonstrated in the target tissues of humans and mice with lupus. In one study, the use of in situ tetramer staining has allowed identification of human antigen–specific T cells in the affected organs, such as the pancreases of patients with autoimmune diabetes.5 Both single and multiple T-cell autoreactivities were detected within individual islets in a subset of patients up to 8 years after clinical diagnosis. Use of such technology has potential to identify tissue-infiltrating autoantigen-specific T cells that can be true targets for treatment in patients with SLE.
Reduced Activation of Regulatory, Inhibitory, or Suppressor T Cells
Immunization with self-Ig peptides induces T cells that can suppress anti-DNA antibodies in healthy strains of mice.45 Induction of such “self-reactive” regulatory T cells is impaired in lupus-prone mice that mount mostly pathogenic T-helper (Th) cell response.
Activation of Toll-Like Receptors
Activation of TLRs by autoantigens can amplify the autoimmune response by activating the innate immune component. Chromatin-containing CpG motif–rich DNA or RNP antigens containing dsRNA can potentially trigger lupuslike autoimmune responses by providing accessory signals through TLR9 on DCs, macrophages, or B cells, or through TLR3 on DCs.46,47 Further, immune complexes containing IgG bound to chromatin can activate murine DCs through both TLR9-dependent and TLR9-independent pathways,48 a feature that may affect autoimmune responses. Indeed, TLR9 deficiency specifically reduces the generation of anti-dsDNA and antichromatin autoantibodies in MRL-lpr mice.49 Viral dsRNA can also activate DCs via TLR3 to induce the production of IFN-I and cytokines associated with disease activity in SLE. Furthermore, TLR3 expression is increased in infiltrating APCs as well as in glomerular mesangial cells in kidney sections of MRL-lpr mice.50
HMGB1, a nuclear DNA-binding protein, can trigger a proinflammatory response by interacting with receptors TLR2, TLR4, and RAGE on macrophages and DCs.51 HMGB1 can also stimulate innate immune responses by acting as a universal sentinel for nucleic acids and facilitating their interaction with a number of receptors, including TLR3, TLR7, and TLR9.52 Importantly, in patients with SLE, serum HMGB1 and anti-HMGB1 autoantibody concentrations are elevated and correlate with disease activity.53 A number of reports have linked the activation of TLRs with autoimmune diseases primarily through their ability to drive the induction of autoreactive T and B cells.54 Furthermore, there is evidence that TLR activation can block T-regulatory (Treg) cell responses, thereby breaking tolerance to self-antigens.
Autoantigens as Chemoattractants
Autoantigens may serve as chemoattractants that recruit innate immune cells to sites of tissue damage.55 A variety of autoantigens has been shown to induce leukocyte migration by interacting with various chemoattractant Gi protein–coupled receptors (GiPCRs). For instance, myositis autoantigen, histidyl-tRNA synthetase, are chemotactic for the CCR5 and CCR3, thereby recruiting T lymphocytes and immature DCs. Fibrillarin (U3-RNP) and topoisomerase I, which are autoantigens associated with scleroderma, have been shown to serve as chemoattractants for monocytes. In some cases, such as in SLE, a complex of two autoantigens has been found to be chemotactic for immature DCs. Thus, autoantigens not only may attract immune cells to a given tissue but also can activate B cells,56 DCs,57,58 and neutrophils59 via their ability to interact with cell membrane receptors.
Altered Recognition of Autoantigens
Altered recognition of autoantigens in endomembrane traffic might elicit autoimmunity.60 The RNA transcription termination factor La, a frequent target of Sjögren autoantibodies, appears in the acinar cell cytoplasm and plasma membranes during viral infection and after in vitro exposure to cytokines. The endomembrane compartments where proteolysis occurs contain La, galactosyltransferase, cathepsin B, and cathepsin D. MHC class II molecules cycle through this compartment. This traffic may permit trilateral interactions in which B cells recognize autoantigens at the surface membranes, CD4+ T cells recognize peptides presented by MHC II, the B cells provide accessory signals to CD4+ T cells, and CD4+ T cells provide cytokines that activate B cells.
Autoantigen Ro52
Autoantigen Ro52 is an E3 ligase that may regulate proliferation and cell death. Increased apoptosis in patients with SLE may result in greater expression of intracellular autoantigen Ro52, which may mediate ubiquitination in survival genes induced during CD40-mediated activation.61 Ro52 may also enhance functions of genes mediating apoptosis and cell death by relieving them from endogenous repression. Intriguingly, Ro52-deficient mice develop autoimmunity, suggesting that this E3 ligase may also act as a negative regulator.
Common Autoantigens in Lupus
Autoimmunity in SLE is directed to some highly conserved intracellular molecules particularly against nuclear and cell membrane phospholipid components.14 Nuclear antigens include DNA-associated autoantigens such as nucleosome and HMGB1 and RNA-associated autoantigens such as U1 snRNP, Ro/La complex, and ribosomes.7 Most studies have focused on autoantibody responses to functionally related nucleic acid–containing macromolecules such as chromatin and RNP particles, because autoantibodies to dsDNA and Sm antigens of the U-1 snRNP complex are considered pathognomonic of SLE. These and other autoantibodies are described in detail in other chapters.
In brief, high-affinity antibodies to dsDNA are hallmarks of SLE. Some subsets of these autoantibodies cause renal and vascular injury.62,63 The common features of such pathogenic autoantibodies, such as class-switched IgGs and somatic mutation, indicate that anti-dsDNA antibodies arise as a result of an antigen-driven process. The antigenic stimuli driving the production of anti-dsDNA autoantibodies remain elusive, but some studies provide possible candidates. Because nucleic acids are poor or not immunogenic, DNA-binding protein in complex with DNA is purported to break tolerance to DNA.64 One possible explanation is that some peptides can serve as surrogate anti-dsDNA epitopes, thus activating T-cell help for the production of anti-dsDNA antibodies.65,66 Another possibility supports a hapten carrier–like mechanism, in which T cells specific for peptides derived from the DNA-binding proteins (such as histones) provide help to DNA-specific B cells. For example, immunization of animals with DNA-protein complexes, rather than with protein-free DNA, induces robust anti-dsDNA antibody response.64 A third possibility is that anti-dsDNA antibody response could occur during the autoantibody response toward the protein constituent of the RNP autoantigens such as nucleosomes or snRNPs.14
Autoantibody against the Sm autoantigens of the snRNP complex is also pathognomonic of SLE. The snRNPs are ubiquitous self-antigens that are components of the spliceosome complex that normally functions to excise intervening introns and generate mature mRNA transcripts. In most snRNP particles, seven core proteins—B, D1, D2, D3, E, F, and G—form a heptamer ring, with the snRNA passing through the center. The Sm epitopes are distributed on the outside surface of the ring. A previous study used overlapping octapeptides spanning the full length of the B/B′ protein to identify an epitope, PPPGMRPP, within the C-terminus of SmB′/B, that is recognized very early in animal models and in some patients with SLE.67 Over time, the immune response spreads beyond this initial epitope to other snRNP autoantigens, including U1-specific RNP epitopes frequently targeted by antibodies present in patients with mixed connective tissue disease.67 Most Sm-precipitin–positive lupus sera, however, recognize certain Sm-D polypeptides, such as the glycine-arginine (GR)–rich carboxyl region of Sm-D1.68,69 The levels of the anti–Sm-D83-119 strongly correlate (as does that of antinucleosome) with disease activity. High levels of anti-dsDNA and anti–Sm-D183-119 strongly correlate with lupus nephritis.70,71
HMGB1-nucleosome complexes from apoptotic cells activate APCs via TLRs and induce proinflammatory responses. HMGB1 is a ubiquitously expressed, structural chromosomal protein that is highly conserved across species.72 In the cell nucleus, HMGB1 binds indiscriminately to the minor groove of DNA and induces strong bends. In addition, HMGB1 is able to bind to highly structured noncanonical or damaged DNA and participates in DNA-related processes, including DNA repair, chromatin remodeling, and transcription. HMGB1 facilitates the formation of multiple nucleoprotein complexes by protein-protein interaction. Initial studies demonstrated the prevention of HMGB1 release during early stages of apoptotic cell death. Hypoacetylation of chromosomal proteins and phosphorylation of histone H2B during apoptotic cell death tightens HMGB1 binding to the chromatin and prevents HMGB1 from being released. By contrast, HMGB1 in necrotic cells is loosely bound to chromatin and is allowed to diffuse into the extracellular space of cells, thereby acting as an endogenous alarmin.73 Evidence now suggests that HMGB1 release also occurs late in the apoptotic cell death process, known as secondary necrosis, in which fragmented HMGB1 is released as a complex with chromatin. Additionally, oxidation of HMGB1 during apoptotic and necrotic cell death may lead to immunogenic neoepitopes that may further exacerbate disease progression.51 HMGB1 itself has been reported to interact with receptors on APCs, including TLR2, TLR4, and RAGE. Interaction with these receptors leads to activation of nuclear factor kappa B (NF-κB), inducing the transcription of proinflammatory genes and the production of inflammatory cytokines. Moreover, HMGB1 induces maturation and migration of DCs. HMGB1 has also been reported to act as a sentinel for virtually all nucleic acids, especially those of viral and microbial origins, thereby aiding in triggering TLR3, TLR7, and TLR9 immune responses by their cognate nucleic acid.52 HMBG1 in complex with nucleosomes from apoptotic cells can activate APCs via a TLR2-dependent pathway, thereby breaking immunologic tolerance against chromatin. Thus, growing evidence suggests a role for HMGB1 in the pathogenesis of SLE.
Some common features of autoantigens are summarized in Box 21-1. In addition to their being evolutionarily conserved and ubiquitously expressed, lupus autoantigens are highly diverse, yet this diversity is restricted to certain sets of autoantigens, causing a “restricted polyclonality” of autoimmune responses in lupus. Several mechanisms have been proposed to explain this phenomenon; they have been reviewed elsewhere.74–76 According to a unique mechanism that we have suggested (Singh et al., 1998b),77 T-cell epitopes (amino acid sequences that can serve as T-cell determinants) are shared among variable regions of different lupus-related autoantibodies but not among other antibodies. Thus, a T-cell epitope present in an anti-DNA Ig may activate T cells that can deliver help to B cells specific for antiphospholipid or anti–red blood cells or other related autoantigens, but not to B cells specific for an unrelated antigen. This is one explanation why lupus autoantibodies are polyclonal yet restricted to a recurring set of autoantigens. The shared T-cell epitopes in autoantibodies may originate as a result of replacement mutations in mutationally “cold” framework regions, which do not affect the binding of antibody to its antigen but create T-cell epitopes.78,79 In fact, although mutations in normal Ig involve hotspot areas, mutations in lupus Ig occur in non-hotspot areas, which might be responsible for creating T-cell epitopes.80
The lupus autoantigens are presumed to initiate and perpetuate the autoimmune response in T and B cells, but exactly how and when this occurs are still not understood. The mechanisms are discussed in other sections of this and other chapters. In brief, autoreactive T cells such as nucleosome-specific T cells have been identified in patients with SLE that drive the formation of anti-dsDNA and antihistone antibodies.81,82
Identification of Autoantigenic Epitopes in Lupus
Studies in Animal Models
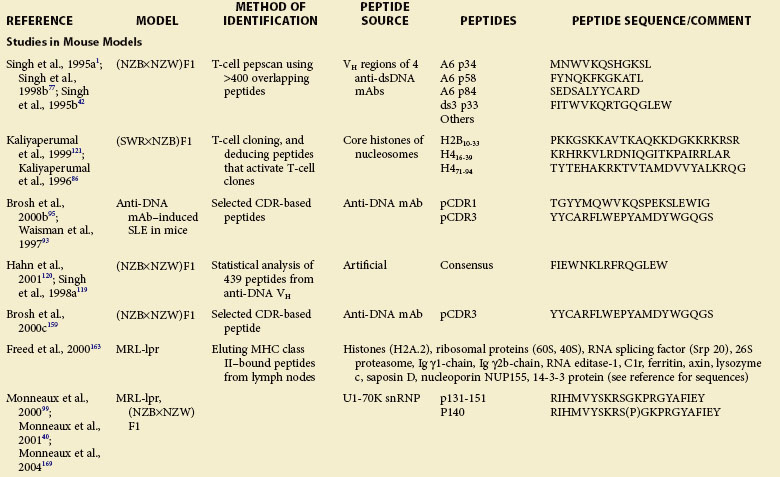
Work in the late 1980s suggested that autoantibody production in humans and mice with SLE is antigen-driven and depends on Th cells that are mostly CD4+.36,83–85 To identify the nature and specificity of such autoreactive Th cells, several laboratories have used diverse approaches, including T-cell pepscanning of candidate autoantigens, isolating autoreactive T-cell clones and deducing potential autoantigens, screening phage display libraries, and eluting naturally processed self-peptides from MHC class II molecules. These approaches have led to identification of epitopes that activate autoreactive Th cells in humans and mice with lupus and modulate disease in lupus mice (see Table 21-1).
Nucleosome Core Histone Peptides as Th Autoepitopes
Using lupus-prone SWR/NZB F1 mice, one group cloned Th cells that can initiate and sustain the production of pathogenic autoantibodies and induce lupus nephritis, and recognize nucleosomes.39 Stimulation of these autoreactive lupus Th cells with 145 overlapping peptides spanning the four core histones H2A, H2B, H3, and H4 led the researchers to localize the critical lupus epitopes in the core histones of nucleosomes at amino acid positions 10 through 33 of H2B and 16 through 39 and 71 through 94 of H4 (see Table 21-1).86 Autoimmune T cells of SWR/NZB F1 mice are spontaneously primed to these epitopes from early life. Moreover, immunization of preautoimmune SWR/NZB F1 mice with these peptides precipitates lupus nephritis.86
In another study, a panel of overlapping peptides spanning the whole sequences of H4 and H3 were cultured with CD4+ T cells from unprimed (NZB/NZW)F1 lupus mice.87 None of the 11 H4 peptides stimulated CD4+ T cells in these mice, whereas several H3 peptides representing sequences 53 through 70, 64 through 78, and 68 through 85 elicited proliferation and induced secretion of IL-2, IL-10, and IFN-γ. The H3 peptides 56 through 73 and 61 through 78 induced the production of IFN-γ and IL-10, respectively, without detectable proliferation, suggesting that they may act as partial agonists of the TCR. Moreover, the study found that this conserved region of H3, which is accessible at the surfaces of nucleosomes, is targeted by antibodies from (NZB/NZW)F1 mice and patients with lupus, and contains motifs recognized by several distinct HLA-DR molecules. This region might thus be important in the self-tolerance breakdown in lupus.
Pulsing bone marrow–derived DCs with lupus autoantigens U1A protein88 or nucleosome89 and then testing in vitro recall T-cell responses to individual epitopes was found to be highly efficient for mapping T-cell epitopes using freshly isolated T cells from unprimed (NZB/NZW)F1 mice. Several potential auto–T-cell epitopes of core histone proteins (H2A, H2B, H3, and H4) were identified with use of this approach.
Self-Ig Peptides as Autoantigenic Epitopes
Early work in the late 1980s and early 1990s suggested that human or murine B cells can process Ig molecules and present Ig-derived peptides in the context of their surface MHC class I and class II molecules. Moreover, Ig-derived peptides are eluted from MHC class II molecules, suggesting that they are naturally processed. T cells from mice expressing a transgene encoding a TCR specific for an Ig-derived peptide provided help for B-cell production of antibodies. Furthermore, Ig peptide–reactive T cells follow rules of conventional T-cell tolerance and activation. It is believed that normal as well as lupus-prone mice generally attain T-cell tolerance to germline-encoded antibody sequences78,90,91 whereas somatically mutated antibody sequences can activate T-cell help because they arise in rare B cells at a late stage of T and B differentiation, thus creating neoepitopes.78 The preceding observations led our group to postulate that SLE B cells process their endogenous or surface Ig into peptides that are presented on MHC class II molecules. These peptide-MHC complexes then activate autoreactive Th cells, which, in turn, stimulate B cells for the increased production of autoantibodies.1,42,77,92
Several lines of evidence support the role of these peptides in autoantibody production and lupus. First, many peptides increased anti-dsDNA antibody production in vitro when cultured with syngeneic splenocytes.77 Second, adoptive transfer of peptide-specific T cell lines or immunizations with peptide/adjuvant emulsions raised serum IgG anti-dsDNA antibody levels, accelerated nephritis, and decreased survival.42
SLE-like disease can be induced in normal mice by injecting human or murine anti-DNA monoclonal antibodies that bear a 16/6 idiotype (Id) that is frequently present on Ig of mice and humans with SLE. Using this model, a previous study found that two peptides representing regions of FR1/CDR1/FR2 (termed pCDR1) and FR3/CDR3 (termed pCDR3) of the heavy chain region (VH) of a monoclonal antibody (mAb), 5G12, stimulated T-cell proliferation in BALB/c and SJL mice and induced proteinuria, leukopenia, and glomerular Ig deposits (see Table 21-1).93 A T-cell line reactive with pCDR3 also induces experimental lupus in naïve mice.94,95 These findings indicate an important role for Ig-derived peptides in the development of lupus.
T-Cell Pepscan of U1-70K snRNP Autoantigen
Muller and colleagues tested a series of overlapping peptides recapitulating the sequence of spliceosomal U1-70K snRNP and identified an epitope present in residues 131 through 151 that is recognized very early by IgG antibodies and CD4+ lymph node T cells in lupus-prone MRL-lpr and (NZB/NZW)F1 mice.96–98 The ability of this peptide to stimulate T cells from mice bearing different MHC haplotypes (H-2k of MRL-lpr and H-2d/z of [NZB/NZW]F1) correlated with its binding to I-Ak, I-Ek, I-Ad, and I-Ed murine MHC molecules. Interestingly, an analog of peptide 131 through 151 sequence phosphorylated on Ser140 (named peptide P140) was more strongly recognized by lymph node and peripheral CD4+ T cells and by IgG antibodies from MRL-lpr mice than the native peptide.75,99 Subcutaneous administration of P140 in Freund adjuvant accelerated lupus nephritis, demonstrating the pathogenic role of a posttranslationally modified epitope.
Screening Phage Display Library to Identify Peptidomimetics That Bind Anti-DNA Antibody
An entirely different approach was used by Gaynor to identify nephritogenic peptides.100 The group screened a peptide display phage library with mouse monoclonal antibodies that bind dsDNA and cause nephritis, and identified a decapeptide DWEYSVWLSN that specifically binds an anti-dsDNA monoclonal antibody, R4A. Immunization with this peptide induced IgG antibodies that bind DNA, cardiolipin, and Sm/RNP and caused Ig deposition in glomeruli.66,101
Identification of Self-Epitopes in Human SLE
Human T cells reactive with several lupus autoantigens, including DNA-histones, the snRNP antigenic proteins Sm-B, Sm-D, U1-70kD, and U1-A, and heterogeneous RNP (hnRNP) A2, have been isolated from the peripheral blood of patients with SLE.102 Datta’s group first described T-cell lines from patients with SLE, which augmented the production of IgG anti-DNA antibodies ex vivo103 and antihistone.104 These autoantibody-promoting T cells are usually CD4+ T cells that use restricted CDR3 characteristic of antigen selection.104 To identify antigenic epitopes for these T cells, they used 154 peptides spanning the entire length of core histones of nucleosomes to stimulate an anti-DNA antibody–inducing Th clone, CD4+ T cell lines, and freshly isolated T cells in peripheral blood mononuclear cells (PBMCs) from 23 patients with SLE.105 In contrast to normal T cells, lupus T cells responded vigorously to certain histone peptides, irrespective of the patient’s disease status (see Table 21-1). Interestingly, most of the peptides that activated human T cells from patients with lupus were previously identified as T-cell epitopes in lupus-prone mice (see Table 21-1). Several additional epitopes, including peptides 34 through 48 of H2A, 91 through 105 and 100 through 114 of H3, and 49 through 63 of H4, were also found to activate human T cells from patients with lupus. Most of these sequences are located in the regions of histones that are accessible at the surfaces of nucleosomes and that contain B-cell epitopes targeted by lupus autoantibodies. Importantly, most T-cell epitopes have multiple HLA-DR binding motifs, that is, they are promiscuous with regard to their binding to HLA molecules. Thus, peptides containing these epitopes could potentially be used to treat many patients, obviating the development of individualized therapy.
To determine whether Ig-derived peptides activate T cells from patients with SLE, Williams cultured PBMCs from 28 patients with lupus and 13 healthy individuals with selected 16-mer peptides from two anti-DNA autoantibodies, B3 and 9G4.106 Three of the 13 healthy individuals (23%) versus 17 of the 28 patients with SLE (61%) had T cells that proliferated in response to at least one V region peptide. In another study, Linker-Israeli cultured 12-mer overlapping peptides from the VH of two anti-dsDNA antibodies, B3 and F51, with PBMCs from patients with SLE, their first-degree relatives, or unrelated healthy individuals.107 The expressions of the early T-cell activation markers CD25 and CD69 and of cytokines were determined by flow cytometry. Patients with SLE had significantly increased T-cell activation markers and IL-4–secreting cells than either first-degree relatives or unrelated controls. A subsequent study by these investigators in a larger cohort (31 patients and 20 matched healthy controls) analyzed cytokine release by PBMCs in response to seven peptides from the CDR1/FR2 to CDR2/FR3 VH regions of human anti-DNA monoclonal antibodies.108 PBMCs from significantly higher proportions of patients with SLE than controls responded to VH peptides by generating IFN-γ and IL-10. Three peptides were more stimulatory in patients with SLE than in controls. There was a skewing of the immune response to Th2 bias as the disease progressed from early to later stage. Although none of the peptides was restricted by any particular MHC class II allele, among “responders” there was greater prevalence of HLA-DQB1*0201 and/or DRB1*0301, alleles known to predispose to SLE. Thus, responses to some VH peptides are more common in SLE and vary with disease duration. Increased peptide presentation by SLE-predisposing HLA molecules might permit brisker increased T-cell responses to autoantibody peptides, thus increasing risk for disease.
Guided by observations in the 16/6 Id murine model described earlier, a previous study examined immune responses of patients with SLE to peptides encompassing complementarity-determining regions (CDRs) of a monoclonal anti-DNA antibody with a 16/6 Id.109 In contrast to the preceding data showing increased responses to anti-DNA–derived peptides,106–108 this group found that peripheral blood lymphocytes (PBLs) from significantly fewer patients (37%) than controls (59%) proliferated in response to one of the anti-DNA peptides.110 A subsequent study by the same group reported in vitro proliferation of PBLs from 24 of the 62 patients with SLE tested after stimulation with the human 16/6 Id.4 Interestingly, peptides from both the human and murine anti-DNA autoantibodies specifically inhibited the 16/6 Id–induced proliferation and IL-2 production. The latter inhibitions correlated with increased production of TGF-β. Findings of this study suggested that certain anti-DNA peptides may downregulate autoreactive T-cell responses in patients with SLE. Indeed, treatment of severe combined immunodeficient (SCID) mice engrafted with PBLs of patients with SLE by repeated intraperitoneal administration of a human monoclonal anti-DNA autoantibody peptide (hCDR1) suppressed human anti-dsDNA antibodies but not anti–tetanus toxoid antibodies.111 Such treatment also reduced proteinuria and renal deposits of human IgG and murine complement C3 in the engrafted SCID mice.
Human T cells reactive with various snRNP antigens, including Sm-B, Sm-D, U1-70kD, and U1-A, have been identified and characterized.102 Subsequent studies on snRNP-reactive human T-cell clones showed that they typically exhibit T-cell receptor alpha/beta–positive (TCRαβ+) CD4+ CD45RO+ phenotype, recognize antigen in the context of HLA-DR, and produce substantial amounts of IFN-γ, moderate quantities of IL-2, and variable amounts of IL-4 and IL-10.112 Further, these cells can also provide help for relevant autoantibody production in vitro.113 Talken established two sets of T-cell clones from patients with connective tissue diseases: one set reacted with Sm-B autoantigen and the others recognized U1-70kD polypeptide.114,115 Both sets of T-cell clones had a highly restricted TCR CDR3 β- or α-chain gene usage, respectively. Further analysis revealed that the Sm-B–reactive T-cell clones recognized a peptide, Sm-B248-96, in the context of HLA-DR. Subsequent T-cell epitope mapping studies of human T-cell clones reactive with the snRNPs U1-70kD, Sm-B, and Sm-D revealed that there are limited T-cell epitopes on these proteins and that almost all reside within functional regions of the protein—either within the Sm motifs for Sm-B and Sm-D or within the RNA binding domain for U1-70kD.
In another study, synthetic 15-mer overlapping peptides spanning the entire La sequence were cultured with PBMCs from patients with SLE and controls with a goal to identify T-cell epitopes in the La antigen. The researchers found a significant, albeit low-level, T-cell proliferative response to a peptide (La 49-63) in HLA-DR3+ patients or healthy controls.116 This study highlights difficulties in identifying relevant pathogenic T-cell epitopes using PBMC-based T-cell proliferation readout experiments. The findings further suggest that the presence of self peptide–reactive T cells in the peripheral blood of healthy individuals is not uncommon. It is unclear why these Th cells promote autoantibody production only in certain individuals.
As described previously, Muller and colleagues identified a CD4+ T-cell epitope in peptide sequence encompassing residues 131 through 151 of the spliceosomal U1-70K snRNP protein (RIHMVYSKRSGKPRGYAFIEY) and its analog phosphorylated at Ser140 (called P140; RIHMVYSKRS(P)GKPRGYAFIEY) in MRL-lpr and (NZB/NZW)F1 mice.40,117 Importantly, administration of the phosphorylated peptide P140 ameliorates the clinical manifestations of treated MRL-lpr mice.40,99 Because this peptide sequence, which is completely conserved in the mouse and human U1-70K protein, contains an RNA-binding motif often targeted by antibodies from patients with lupus and mice, the group investigated these peptides as potential candidates for the treatment of patients with lupus.117 Binding assays with soluble HLA class II molecules and molecular modeling experiments indicate that both peptides behave as promiscuous epitopes and bind to a large panel of human DR molecules. In contrast to normal T cells and T cells from patients with non-SLE autoimmune disease, PBMCs and/or CD4+ T cells from 40% of patients with SLE proliferate in response to peptide 131-151. Interestingly, the phosphorylated analog peptide P140 prevents CD4+ T-cell proliferation but not secretion of regulatory cytokine IL-10. Thus, P140 can serve as a “universal” immunomodulatory T-cell epitope.
Related posts:
 The Innate Immune System in SLE
Clinical Aspects of the Antiphospholipid Syndrome
Gastrointestinal and Hepatic Manifestations
Novel Therapies for SLE: Biological Agents Available in Practice Today
Mixed Connective Tissue Disease and Undifferentiated Connective Tissue Disease
Pathomechanisms of Cutaneous Lupus Erythematosus
The Innate Immune System in SLE
Clinical Aspects of the Antiphospholipid Syndrome
Gastrointestinal and Hepatic Manifestations
Novel Therapies for SLE: Biological Agents Available in Practice Today
Mixed Connective Tissue Disease and Undifferentiated Connective Tissue Disease
Pathomechanisms of Cutaneous Lupus Erythematosus
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


