Chapter 11 Apoptosis, Necrosis, and Autophagy
Definitions
Apoptosis
The modern understanding of apoptosis began with the electron-microscopic descriptions of morphologic changes characterized by shrinkage of hepatocytes (i.e., shrinkage necrosis) after ischemic or toxic injury to the liver. The name apoptosis was coined by Kerr in 1972 to describe the form of death that was “consistent with an active, inherently controlled phenomenon” characterized by cell shrinkage, nuclear condensation, and cell blebbing (Figure 11-1).1 This term also conveyed the concept of cell death that was similar to leaves falling from a tree (apo means “from” and ptosis “a fall” in Greek), implying a regulated “mechanism of cell deletion, which is complementary to mitosis.”1
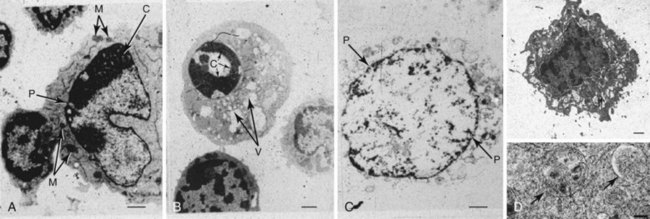
FIGURE 11-1 Electron-microscopic morphology of cell death.
(Autophagy image courtesy of Dr. Mike Lenardo. Adapted from Russell JH, Masakowski V, Rucinsky T, et al: Mechanisms of immune lysis III: Characterization of the nature and kinetics of the cytotoxic T lymphocyte induced nuclear lesion in the target. J Immunol 128:2087, 1982.)
Further developments in our understanding of apoptosis paralleled advances in molecular biology, genetics, and biochemistry. The detection of a nucleosomal ladder2 was of considerable importance, because it defined a biochemical event (i.e., nucleosomal cleavage) and provided a simple electrophoretic test for detection of apoptotic cell death that remains a standard in the field. Studies in the 1980s demonstrated that the death of cells during nematode development was under strict genetic control. Remarkably, the death of these cells could be perturbed by mutation of a small number of genes called ced (for cell death abnormal) genes.3 Horvitz determined that two ced genes, ced3 and ced4, encoded death effectors, whereas ced9 was an antiapoptotic gene. Most of the remaining ced genes were responsible for engulfment and removal of the “corpses.” This simple model, in which CED-3 is the main death protease that is activated by CED-4 and inhibited by CED-9, has served as a paradigm for defining apoptotic pathways in mammalian cells. Mammalian cells are more complex and, as discussed here in detail, have multiple defined pathways that follow the basic Caenorhabditis elegans model. The molecules within these pathways, the downstream effectors of apoptosis, the caspases (cysteine aspartate proteases), and the proteins implicated in the clearance of apoptotic cells are discussed in detail. The control of cell death is of seminal importance in a number of diseases, including cancers, autoimmune diseases, and degenerative disorders.4 Regulation of apoptosis and handling of dying cells are especially relevant to SLE, as also discussed here.
Necrosis
Necrosis has traditionally been viewed as a passive form of cell death resulting from toxic or physical insults leading to adenosine triphosphate (ATP) depletion, although later studies indicate that some changes can be biochemically mediated. Morphologically, necrosis is notable for plasma cell membrane disruption leading to cellular swelling and cytoplasmic vacuolation (see Figure 11-1). In contradistinction to apoptosis, necrotic death induces inflammation around the dying cell as a result of the release of various intracellular components (see Figure 11-1). Although chromatin degradation is evident in necrotic cells, the condensation and organized DNA fragmentation seen in apoptotic cells are usually absent. Notably, the same inducers (e.g., ischemia, hydrogen peroxide) may produce apoptosis or necrosis, depending on the severity of the injury and the rapidity of cell death. The cell’s fate is determined in part by cellular energy reserves such as ATP. When removal of the apoptotic cells is delayed, some inducers initially cause apoptosis followed by necrosis (postapoptotic necrosis). This transition from apoptotic to necrotic cells is likely to be important in the context of autoimmunity (see later).
Autophagy
Autophagy, which means to eat oneself, occurs during nutrient stress when cells switch to a catabolic program and degrade cytoplasmic constituents as a survival mechanism.5 It does not necessarily lead to cell death. During autophagy, more than 20 members of the ATG family of proteins orchestrate the initiation, elongation, and closure of the characteristic double-membrane vesicle called the autophagosome. The autophagosome then fuses with lysosomes (see Figure 11-1), the contents are degraded, and the products recycled for energy utilization. Autophagy has been implicated in a wide spectrum of diseases ranging from degenerative neurologic diseases associated with polyglutamine repeats to cancer and immunologic diseases such as Crohn disease.5
Autophagy is important in immune functions, such as the intracellular host response to pathogens, survival of lymphocytes following growth factor withdrawal, Toll-like receptor (TLR) stimulation in plasmacytoid dendritic cells (pDCs), and major histocompatibility complex (MHC) class II presentation of antigen.5,6 The association between a genetic variant of ATG16L1 as well as IRGM (immunity-related guanosine triphosphatase gene), another protein that regulates autophagy, and Crohn disease is unclear but could potentially involve thymic selection, macrophage function, or intestinal immunity.5 Of relevance to SLE is the identification in patients with the disease of an increased frequency of single-nucleotide polymorphisms) (SNPs) in ATG5 that could potentially predispose to disease through alterations in T-cell selection, clearance of apoptotic cells, or regulation of type I interferon (IFN).5
Other Forms of Cell Death
Pyroptosis (from the Greek, “pyro” meaning fire) is distinguished from the other forms of cell death by the activation of caspase 1 (interleukin-1beta [IL-1β]–converting enzyme) and secretion of the inflammatory cytokine IL-1β. Pyroptosis is most strongly associated with infections by intracellular bacteria such as Salmonella, Yersinia, and Legionella, although it may also be seen following tissue infarction.7 Cells undergoing pyroptosis demonstrate nuclear condensation associated with DNA damage, cell swelling, and, ultimately, cell lysis associated with release of IL-1β. The mechanisms responsible for this process involve intracellular sensors of bacterial products and formation of the inflammasome.
Necroptosis is a programmed form of necrosis that has so far mainly been described in response to certain death ligands such as Fas ligand and tumor necrosis factor alpha (TNF-α; see Biochemistry of Apoptosis). It has long been known that in addition, a stimulation of inflammation through activation of nuclear factor kappa B (NF-κb), TNF-α can induce necrosis (tumor necrosis factor). Only recently has the pathway become clear, and it is described in the section Initiation and Pathways of Apoptosis.
Biochemistry of Apoptosis
Figures 11-2 and 11-3 show schematic overviews of the cell death program. A brief outline of each major functional component within the program, from the signals for death to removal of the apoptotic cells, is discussed here, but space limitations preclude a detailed analysis of the layers of regulation at each step of the pathway. For a more comprehensive discussion of the biochemical pathways controlling apoptosis, the reader is referred to excellent reviews.8,9
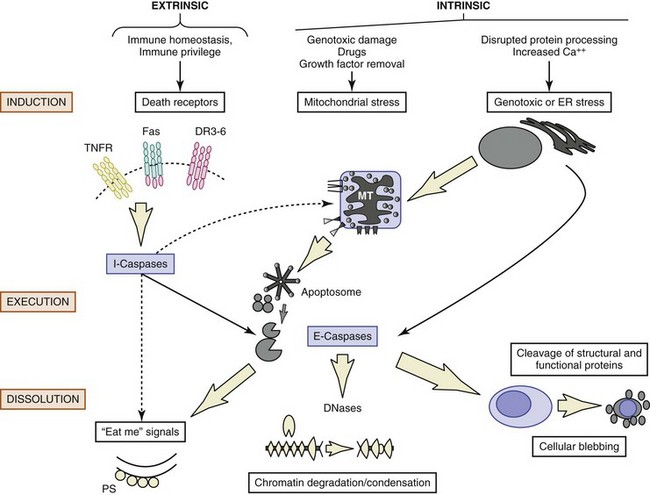
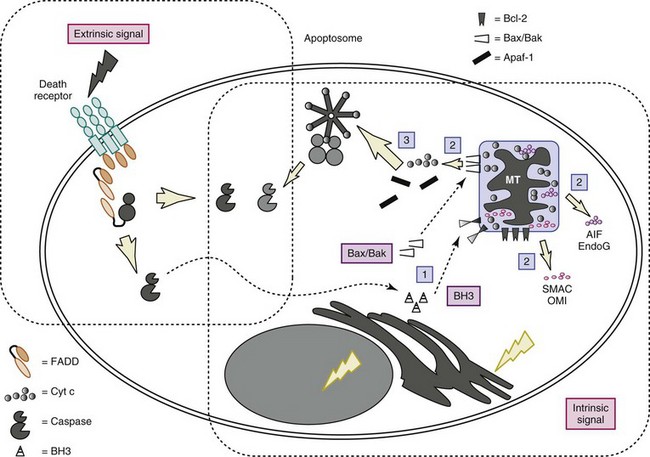
Numerous proteins involved in apoptosis, including receptors, adaptors, effectors, and inhibitors, contain modules/domains that are structurally similar and evolutionarily conserved. Interestingly, these motifs are predominantly involved in promoting homotypic protein-protein interactions (see Figure 11-3). Furthermore, as discussed later, these domains occur in proteins involved in apoptosis as well as inflammation. It has been suggested that death domain (DD), death effector domain (DED), caspase recruitment domain (CARD), and pyrin domains all evolved from the prototypic DD-fold corresponding to an antiparallel six-helix bundle.10 Because of the central role for caspases, deoxyribonucleases (DNases), and the bcl-2 family of proteins, each is briefly described in greater detail later, before the discussion of how these families of proteins interact.
The cell death process can be divided into a number of stages, as follows: inductive stimulus, signal transduction, activation of caspases, activation of nuclease(s) with nuclear condensation, redistribution of the cellular contents into apoptotic bodies, and removal of the dying cells (see Figures 11-2 and 11-3). Because the nature of the inductive stimulus dictates the initial biochemical pathways engaged, extrinsic (“death receptor”) and intrinsic (damage- and stress-induced) pathways are discussed separately.
Caspases
The cysteine protease family of caspases plays a central role in apoptosis, and the orthologs in C. elegans and Drosophila demonstrate evolutionary conservation. Caspases are produced as catalytically inactive zymogens and function as homodimers, with each monomer composed of a large subunit and a small subunit. Caspases can be divided into two categories reflecting both structural and functional differences; “initiator” and “effector” caspases (Table 11-1; for a comprehensive review, see reference 11).
TABLE 11-1 Families of Intracellular Proteins Involved in Apoptosis
| Caspases | ||
|---|---|---|
| Initiator | Pro-Domain | Function |
| Caspase 8, 10 | Long, with DED | Extrinsic (death receptor) pathway |
| Caspase 9 | Long, with CARD | Intrinsic pathway |
| Caspase 2 | Long, with CARD | Both extrinsic (death receptor) and intrinsic (chemotherapy) pathways |
| Executioner | ||
| Caspase 3/6/7 | Short | Cleavage of apoptotic substrates |
| Caspase Inhibitors | ||
| IAPs | — | Active-site blockage of caspases |
| p35, CrmA | — | Pan-caspase inhibitors |
| cFLIP | DED | Cell inhibitor of DISC formation |
| Bcl-2 Members | ||
| Prototype | Others | |
| Anti-apoptosis: | ||
| Bcl-2 sub-family | Bcl-2 | Bcl-xL, Bcl-w, Mcl-1 |
| Pro-apoptosis: | ||
| Bax sub-family | Bax | Bak, Bok |
| BH3-only sub-family | Bid | Bim, Bad, Puma, Noxa, Bik |
| DNases | ||
| DNase | Activation | DNA substrates |
| CAD | Caspase | Cell autonomous, generates nucleosomes |
| AIF + Endo G | Mitochondrial | Cell autonomous, cleaves chromatin |
| DNase II | Low pH | Ingested DNA in phagocytes |
| DNase I | — | Extracellular DNA |
| TREX1 | — | Intracellular single-stranded DNA |
AIF, apoptosis-inducing factor; CAD,; CARD, caspase recruitment domain; cFLIP, cellular FLICE (FADD [Fas-associated protein with death domain]–like interleukin-1β–converting enzyme)–inhibitory protein; DED, death effector domain; DISC, death-inducing signaling complex; Endo G, endonuclease G; IAP, inhibitor of apoptosis; TREX1, three prime repair exonuclease 1.
The “initiator” caspases, which in mammals include caspases 2, 8, 9, and 10, have extended N-terminal prodomains that allow for clustering and autoactivation of the zymogens. The clustering of the initiator caspases depends on adaptor proteins that utilize homophilic interactions between conserved nonenzymatic domains present on both the adaptor and the caspase. These adaptors include Fas-associating protein with death domain (FADD), which recruits caspase 8 via homophilic interactions between DEDs in both proteins (see later and Figures 11-2 and 11-3), and apoptotic protease activating factor 1 (Apaf-1), which recruits caspase 9 through a homophilic interaction in CARD (caspase recruitment domain). The recruitment and activation of various initiator caspases occur in response to unique sets of stimuli and in separate cellular compartments. Autoactivation of initiator caspases likely occurs through oligomerization, ultimately leading to the activation of downstream effector caspases (caspases 3, 6, and 7). As discussed later, caspase 8, considered a pro-apoptotic protease, functions both in Fas-induced apoptosis and in lymphocyte activation and protective immunity.
Not all caspases are involved in the execution of apoptosis. Human caspases 1, 4, and 5 are most likely involved in inflammation. Caspase 1 was originally defined as the enzyme that cleaves IL-1 (interleukin-1–converting enzyme [ICE]) into its active form. It has been shown that caspases 1 and 5 interact to form a multiprotein complex that has been called the inflammasome (analogous to the apoptosome).12 Caspases 1 and 5 bind to the adaptor proteins ASC (pyrin CARD [PYCARD]) and NALP1 (DECAP), respectively, by their CARD domains. The protein, pyrin, which is mutated in familial Mediterranean fever (FMF), binds to ASC. Mutation of pyrin therefore most likely leads to inflammation through uncontrolled activation of caspase 1, generating active IL-1 and IL-18.
Inhibition of Caspases—Intracellular Inhibitors of Apoptosis
Intracellular inhibitors of apoptosis (IAPs) are a family of antiapoptotic proteins that are highly conserved through evolution. Members of the family (cellular IAPs c-IAP-1 and c-IAP-2, X-linked IAP [X-IAP], survivin, IAP-like protein 2 [ILP2], ML-IAP (melanoma IAP), Bruce, and neuronal apoptosis inhibitory protein [NAIP]) share a baculovirus IAP repeat (BIR) domain and most contain a RING domain that functions as an E3 ligase. IAPs such as X-IAP directly inhibit effector caspases, especially caspase 9, whereas c-IAPs modulate cell survival by ubiquitylation of substrates such as ribosome-inactivating protein (RIP) and proteins in the NF-κB pathway. IAPs block apoptosis induced by a variety of stimuli, including Fas, TNF-α, ultraviolet (UV) irradiation, and serum withdrawal. IAPs themselves are inhibited by two mitochondrial proteins named Smac/Diablo and HtrA2/Omi, which are released into the cytosol during the intrinsic and some extrinsic apoptotic programs (see Figure 11-3).
Nucleases and the Degradation of Cellular DNA and RNA
Nucleic acids are of high relevance to SLE as they are prominent targets of autoantibodies in this disease and can potently stimulate the production of type I IFN. RNase 1 and DNase I are the predominant serum nucleases thought to be crucial for the degradation of nucleic acids released from dead and dying cells into blood or tissue. Mice deficient in DNase I, at least on some strain backgrounds, demonstrate a lupus-like phenotype,13 but whether altered DNase activity represents a genetic susceptibility factor in human lupus remains controversial.
A hallmark of programmed cell death is internucleosomal cleavage of DNA, which results in nuclear condensation and a nucleosomal “ladder.” Numerous DNases have been implicated in this process, including caspase-activated DNase (CAD/DFF40) and apoptosis-inducing factor (AIF)/endonuclease G (Endo G) (located in mitochondria) (see Table 11-1). Initially, degradation of chromatin into large DNA fragments to produce large (50-200 kb) fragments occurs through cooperative interaction between endo G and AIF. Subsequently, while the cell membrane remains intact, CAD/DFF40 cleaves DNA into internucleosomal units, producing a characteristic 180-bp ladder. Following phagocytic ingestion of apoptotic cells with partially processed DNA, DNase II, another DNase located in lysosomes, completes DNA digestion. Mice deficient in both CAD/DFF40 and DNase II have defects in thymic development associated with production of inflammatory cytokines, supporting a model in which DNA degradation during apoptosis is a sequential process that requires initiation in a cell-autonomous way and is then fully executed by the phagocyte. Of interest, mice doubly deficient in DNAseII and IFN receptors demonstrate a disease similar to rheumatoid arthritis.14
A third category of nucleases are those involved in processing of normal or abnormal (e.g., virus) nucleic acids. TREX1 (three prime repair enxonuclease 1) is a 3-5′ exonuclease that appears to be necessary for degradation of cytoplasmic single-stranded DNA.15 Loss of TREX1 function stimulates the production of type I IFN through an unknown DNA sensor.
The Bcl-2 Family: Central Regulators of Apoptosis
There are at least 20 known Bcl-2 family members in mammals that have diverse cellular localization and function and are broadly divided into three interacting groups (see Table 11-1; and reviewed in reference 16). All Bcl-2 family members possess one or more BH (Bcl-2 homology) domains, which allow for interaction with a variety of proapoptotic or antiapoptotic proteins. Multidomain prosurvival members Bcl-2, and closely related Bcl-xL, Mcl-1, and A1, can protect cells from a wide range of potentially death-inducing stimuli, including irradiation (UV and gamma), growth factor withdrawal, and chemotherapy. The proapoptotic multidomain group that includes Bax, Bak, and Bok likely functions by altering the permeability or conductance of mitochondrial and other membranes, resulting in the release of additional proapoptotic mediators (see later). Finally, the “BH3-only” group of proapoptotic proteins contains at least eight members and functions primarily by antagonizing the protective effects of the Bcl-2 family. Some BH3-only proteins may sensitize cells for apoptosis, whereas others likely function more directly in activating apoptosis.8 The BH3-only members act as sentinels in various organelles, integrating proximal death or survival signals and ultimately facilitating Bax/Bak-induced apoptosis.
Abnormalities in the Expression of Bcl-2 Family Members Cause Lupus-Like Autoimmunity in Mice
Mice that overexpress the antiapoptotic protein Bcl-2 or are deficient in the proapoptotic protein Bim demonstrate a lupus-like disease on certain strain backgrounds.17 Bcl-2 is not a critical player in positive or negative thymic selection but may promote autoimmunity by enhancing cell survival in peripheral lymphocytes. In contrast, loss of Bim does affect thymic selection, as evidenced in a Bim-deficient T-cell receptor transgenic model in which T cells targeted against the male antigen HY have impaired deletion of autoreactive CD4+8+ thymocytes in male mice.18 Bim may also regulate B-cell survival.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


