Chapter 17 Animal Models of SLE
Animal models of SLE have provided an invaluable resource for defining disease pathophysiology, genetics, and therapeutic approaches. Furthermore, in human SLE, heterogeneity in disease expression is an obstacle to devising targeted interventions that apply to all patients. Consequently, many investigators have turned to murine lupus models, both spontaneous and induced, that develop relatively homogeneous disease recapitulating some of the serologic and histopathologic features of SLE. Murine SLE is characterized by the presence of autoantibodies against nuclear and a variety of other ubiquitous and cell type–specific self-antigens, and end-organ injury, most commonly immune-mediated glomerulonephritis (GN).1 Another noteworthy finding is that virtually all cases of spontaneous and induced murine lupus require a susceptible genetic background. There are informative differences in disease in different strains. For example, BALB/c mice injected with a DNA surrogate peptide demonstrate extensive glomerular immune deposition but no renal inflammation,2 whereas in BALB/c mice injected with hydrocarbon oil pristane immune deposition and limited kidney inflammation (focal GN) develop, but no kidney failure.3,4 In contrast, many multigenic lupus-prone mouse strains spontaneously develop immune deposition–mediated renal disease that is lethal, with strain-dependent variation in disease patterns and severity.5 Numerous single-gene knockout and transgenic strains also develop autoimmune features6,7 that recapitulate some aspects of SLE-like disease.
A few lupus-like murine strains develop coronary occlusions and myocardial infarction as a result of immune complex deposition, but the relevance of this mechanism to the increased cardiovascular disease in human SLE is unclear. Some strains develop dermatitis, autoimmune hemolytic anemia, arthritis, and vasculitis, but the incidence of these disorders is generally variable and may depend at least in part on environment. Mouse models have not recapitulated the waxing and waning nature or the full spectrum of human SLE. Spontaneous SLE also develops in dogs. Tables 17-1 to 17-3 provide an overview of major characteristics of mice and dogs with lupus-like disease.
TABLE 17-1 Major Characteristics of Animal Strains Developing Systemic Lupus Erythematosus (SLE): Disease Manifestations in Lupus-Prone Animals
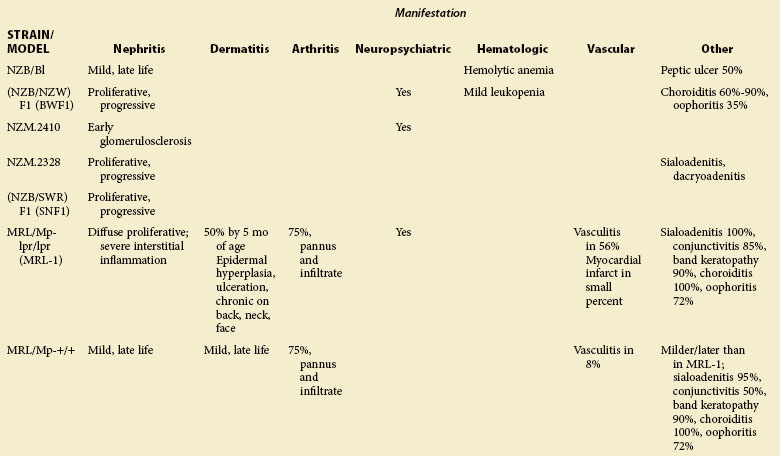

TABLE 17-2 Major Characteristics of Animal Strains Developing Systemic Lupus Erythematosus (SLE): Autoantibodies in Lupus-Prone Animals*
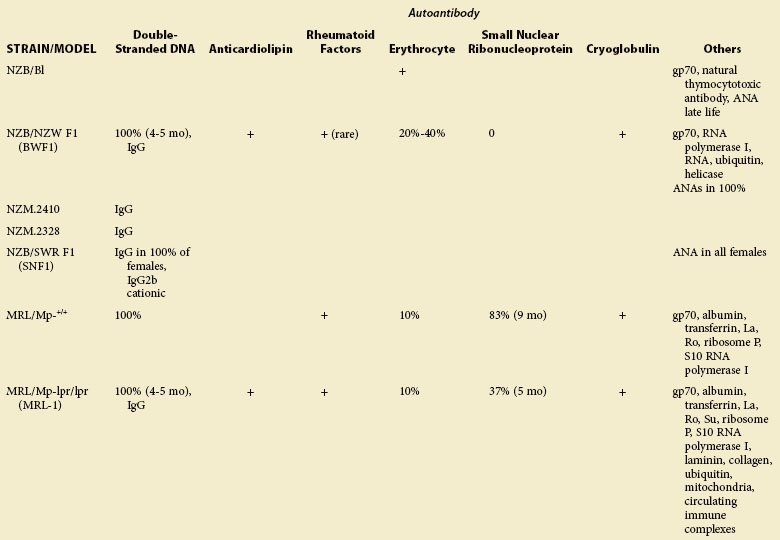
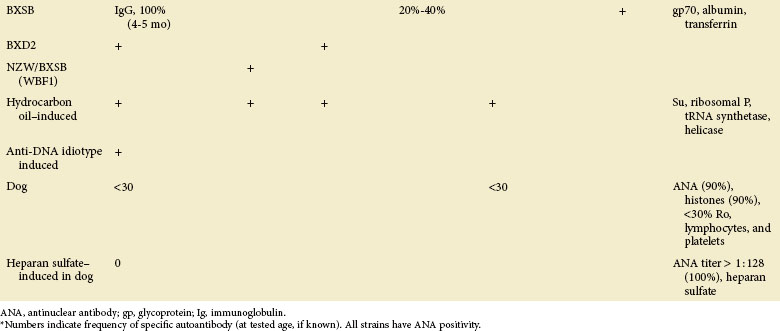
TABLE 17-3 Major Characteristics of Animal Strains Developing Systemic Lupus Erythematosus (SLE): Genetic and Other Features of Lupus-Prone Animals
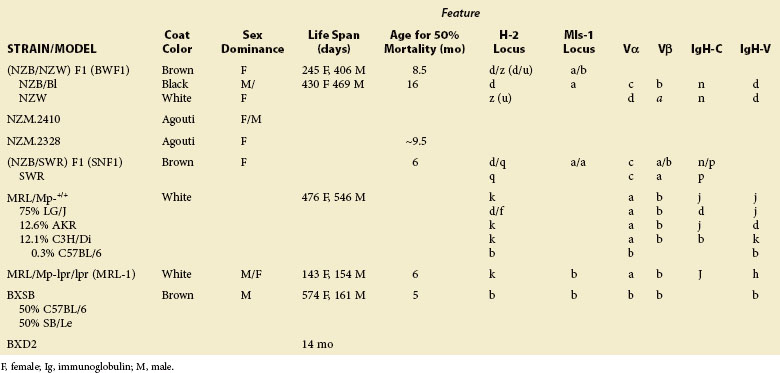
Clinical Disease, Autoantibodies, Immunologic Abnormalities, and Genetics in Spontaneous Multigenic Murine SLE
New Zealand Mice
NZB/BL (NZB) Mice
The New Zealand Bielschowsky black (NZB/Bl) mouse was bred by Bielschowsky, who was mating mice by coat color to derive cancer-susceptible strains. In 1959, she reported that NZB mice died early from autoimmune hemolytic anemia.8 Shortly thereafter, her colleagues described a hybrid between NZB and unrelated strains including the New Zealand white (NZW) that was characterized by early death in females from nephritis associated with lupus erythematosus (LE) cells, thus providing the first animal models of lupus nephritis.9
The characteristics of NZB mice are shown in Tables 17-1 to 17-3 and Box 17-1. They also are discussed in several review articles.10,11
Box 17-1
Characteristics of NZB/BI Mice
Immune Abnormalities
1. B cells are unusually mature and hyperactivated, and they secrete Ig spontaneously from a very early age (in fetus and in newborn mice); this abnormality is required for autoimmune disease in NZB mice and in hybrids mated with NZB mice.
2. Numbers of B-1 (CD5+) B cells in spleen and peritoneum are increased; these cells make primarily IgM autoantibodies; their elimination protects from SLE.
3. B cells resist tolerance to T cell–independent antigens.
4. Older mice develop aneuploidy in B-1 B cells.
5. Thymic epithelium is strikingly atrophic by 1 month of age.
6. Antithymocyte antibodies react with immune T cells and may inactivate/delete precursors of suppressor T-cell populations.
7. T cells are required for maximal autoantibody formation.
8. High quantities of a unique form of retroviral gp70 antigen in serum.
9. Clearance of immune complexes by Fc-mediated mechanisms is defective.
Genetics
1. Multiple dominant, codominant, and recessive genes participate in the immune abnormalities.
2. One set of genes controls the constellation of polyclonal B-cell activation, expression of gp70, and antithymocyte antibodies; another set of genes controls B-cell tolerance defects, antibodies to gp70, anti-ssDNA, and anti–red blood cells; the gene sets segregate independently; neither of these sets is dependent on H-2.
3. The disease is linked to major histocompatibility class.
4. NZB has two to five susceptibility genes located on different chromosomes, some transmitted in a dominant and others in a recessive fashion. Nba2 region on chromosome 1 plays a major role in susceptibility to lupus in mice with NZ backgrounds.
gp, glycoprotein; Ig, immunoglobulin; ssDNA, single-stranded DNA.
Clinical Characteristics and Autoantibodies
NZB mice are characterized by hyperactive B cells, present in fetal life, that produce primarily immunoglobulin M (IgM) antibodies to thymocytes, erythrocytes, single-stranded DNA (ssDNA), and the gp70 glycoprotein of murine leukemia virus.10–12 The first antibody to appear in serum is natural thymocytotoxic antibody (NTA)13,14; by 3 months of age, 100% of mice have this antibody. NTAs are cytotoxic for all thymocytes, 50% to 60% of thoracic duct and peripheral blood lymphocytes (both CD4+ and CD8+ populations), 50% of lymph node cells, 33% of spleen cells, and 5% of bone marrow cells. These figures are similar to the reactivity of anti–Thy-l sera. Some NTAs react with cell surface molecules on B lymphocytes, granulocytes, and bone marrow myeloid cells; others react with a 55-kd molecule on most T cells. Other reported reactivities include an 88-kd glycoprotein, which is thought to be a T-cell differentiation antigen, and surface molecules of 33 and 30 kd.11,15–17
The primary clinical problem in NZB mice is hemolytic anemia, which is fatal in most (60%-90%) between 15 and 18 months of age.8,10,11 There is mild disease acceleration in females, with death 1 month earlier than in males. IgM and IgG antibodies to erythrocytes (red blood cells [RBCs]) cause hemolysis,18,19 appear by 3 months of age, and are found in 100% by 9 to 10 months of age. Antibodies to RBCs are directed against (1) RBC surface antigens exposed by bromelain, (2) anion exchanger membrane protein band 3,20–22 or (3) spectrin.23 Early in life, the RBC antibodies are polyreactive; later, they become more specific for band 3 or spectrin, suggesting antigenic stimulation.23 However, genetic deletion of the band 3 antigen does not protect NZB mice against anti-RBC: they simply make autoantibodies to antigens other than B and 3.21 Polyclonal B-cell activation, B-cell proliferation, high IgM production, and class-switching in response to activation by Toll-like receptors (TLRs) are largely independent of T-cell help and may be influenced by elevations of B cell–activating factor (BAFF) in this strain.24 However, hemolysis is primarily mediated by IgG anti-RBC, and the characteristic mild clinical GN25,26 depends on deposition of IgG autoantibodies (particularly to nucleosome and dsDNA), especially of the T helper–1 cell (Th1)–driven, IgG2a complement–fixing subclass. Knockout of CD40L (which mediates 2nd signal B-T interactions that result in Ig class switch) abrogates most IgG autoantibody production, and largely prevents glomerular disease, but does not affect abnormalities in B220+ cells and in IgM production.24 In NZB mice, antinuclear antibodies (ANAs) are not regularly present in high titers as in other lupus-prone strains, although approximately 80% of mice test ANA positive by 9 months of age.10 Some NZB mice exhibit learning disabilities,27 probably related both to the cortical ectopias that occur in 40% and to the autoimmune process. Autoantibodies to Purkinje cells of the cerebellum have been found,28 and the numbers of interleukin-1 receptors (IL-1Rs) expressed in the dentate gyrus are lower than in normal mice.29
Abnormalities of Stem Cells and B Cells
NZB mice are remarkable for inherent abnormalities in their B cells that probably originate in bone marrow stem cells, because hyperactivation of B cells is detectable in fetal liver. In comparison with normal mice, there are higher numbers of IgM-secreting cells and greater synthesis of IgM by individual B cells—characteristics that may be controlled by different genes.30–32 Splenic CD21hiCD23− (marginal zone [MZ] B cells) and CD21loCD23− (immature B, memory B, and preplasma cells) subsets are particularly expanded, as are spleen and bone marrow plasma cells.24 Putative bone marrow pre-B cells exhibit increased growth both in vitro and in vivo33; this property is lost after 10 months of age.34 Mature B cells are resistant to normal control mechanisms involving engagement of the B-cell receptor (BCR).35 Another B-cell abnormality highly characteristic of NZB mice is the appearance of aneuploidy in B cells, primarily in B-1 B cells (also designated Ly-1 or CD5+), as the mice age. Hyperdiploid B-1 B cells with additional chromosomes 10, 15, 17, and X are common.36 Lymphoid malignancies are more common in NZB than in other murine lupus strains, prevalence varying in different colonies between 1% and 20%; they may be a model of B-cell chronic lymphocytic leukemia. Malignant B-1 B cells secrete large quantities of IL-10, which can skew T-cell repertoires away from T-helper 1 (Th1) and toward Th2 phenotypes.37 In young NZB mice, numbers of nonmalignant B-1 B cells are increased in the spleen and peritoneum38; these cells make IgM autoantibodies to RBCs, thymocytes, and ssDNA. B-1 cells are also present among MZ B cells in lymphoid tissues.39 B-2 (CD5−) B cells, however, are more likely to be the source for IgG autoantibodies.40 Nevertheless, elimination of B-1 B cells by lysing of the cells with water in the peritoneal cavity (where these cells are renewed) reduces antibodies to RBC and hemolytic anemia,41 thus demonstrating the importance of B-1 cells to NZB disease. Finally, splenic B cells in NZB mice are probably resistant to apoptosis because of the influence of the Ifi202 gene (in the Nba2 region), which is upregulated in this strain and plays a major role in sustained autoantibody production in NZB hybrids.42,43
Abnormalities of Dendritic Cells
Since the recognition of the connections between innate and acquired immunity, there has been great interest in the role of dendritic cells (DCs) as mediators of immune tolerance, as a source of antigen-presenting cells (APCs) that activate T cells, and as a source of interferon alpha (IFN-α). Notably, pDCs express TLR9 and TLR7, which can bind immune complexes containing, respectively, DNA and ssRNA, thus becoming activated pDCs that enhance autoimmune responses to nucleic acids or material containing nucleic acids. In fact, NZB mice, compared with normal strains, respond to injections of CpG oligodeoxynucleotide (CpG ODN), a synthetic DNA, with increased release of IFN-α. Furthermore, cell numbers of DCs and messenger RNA (mRNA) for TLR9 are increased in NZB mice. On the other hand, other features of DCs that promote inflammation are abnormally low in NZB DCs, including production of IL-12 and expression of the homing chemokine CCR7 and the activation surface marker CD62L.44 Whether these DC abnormalities represent primary defects contributing to autoimmunity or secondary activation stages remains to be determined.
Abnormalities of Thymus and T Cells
NZB mice characteristically exhibit a dramatic involution of thymic tissue; thymic epithelium is atrophied and immunologically defective by 1 month of age (before the appearance of NTA), with epithelial cell degeneration, accumulation of terminal deoxynucleotidyl transferase-positive (TdT+) large immature T cells in the subcapsular region of the cortex, cortical atrophy, and increased lymphoid and plasma cell infiltrates in the medulla.10,17,45–47 NZB thymic epithelial cells are functionally defective compared with cells from normal mice, having low expression of Aire and RelB proteins, low numbers of surface Ia molecules and major histocompatibility complex (MHC) class II molecules, low secretion of IL-1, high secretion of prostaglandin E2 (PGE2) and PGE3, and diminished ability to educate nonthymic cells to express Thy-l.46–49 NZB bone marrow contains greater prothymocyte activity, and the prothymocytes have an increased growth advantage when they are transferred to histocompatible recipients.50 Thymic DCs are also abnormal; they are defective in mediating negative T-cell selection, possibly because they express less E-cadherin than thymic DCs from nonautoimmune mice.51
CD4+ T cells play an essential role in NZB disease. Accordingly, the MHC class II (also called H-2 in mice) is an important predisposing factor for autoimmunity. The hybrid combination of NZB (H-2d/d haplotype) and NZW (z/z) or SWR (q/q) to make d/z or d/q MHC haplotypes enhances susceptibility to GN mediated by IgG anti-dsDNA,52–62 which are antibodies that NZB mice do not make. NZB mice that are congenic for H-2b (NZB.H-2b) have less disease than the wild-type NZB.H-2d. However, introduction of a mutated I-A chain (bm12) converts this animal (i.e., NZB.H-2bm12) to a phenotype that is similar to the BWF1 hybrid, with high-titer IgG anti-dsDNA and severe clinical GN.63,64 MHC class II likely plays a role in disease by shaping the repertoires of CD4+ T cells. In fact, CD4+ cells that proliferate in response to the RBC membrane protein band 3 and to spectrin have been isolated from NZB spleens.22 The importance of T cells to autoantibody formation is also indicated by experiments in which anti-CD4 nondepleting antibody was administered to NZB mice; antierythrocyte antibodies were significantly decreased, although anemia was not prevented.65
Genetics
Genetic susceptibility in NZB mice is polygenically inherited, with genes having a partial and additive effect similar to that in most human SLE. With use of crosses of NZB to lupus-prone and non-lupus strains, loci linked to one or more lupus traits have been mapped to ten chromosomes.52–62 The underlying genetic variants for most loci are not yet defined. For the most prominent disease manifestation, anti-RBC antibodies, loci on chromosomes 1 (called Nba2), 4 (Aia1, Aem1), 7 (Aem2), and 10 (Aem3) have been confirmed by more than one study or in congenic mice.57,63–67 Among these, the distal chromosome 4 locus (also called Lbw2) was further dissected with subcongenic mice and shown to consist of at least three loci.66 This locus also increased susceptibility to GN and B-cell hyperactivity on the BWF1 background.68 IgM hypergammaglobulinemia and increased peritoneal B-1 cells also map to the same interval.36,60,69,70 Within this interval, a promoter variant of Cdkn2c (encodes the cyclin-dependent kinase inhibitor p18INK4c) associated with reduced expression in a B6-Sle2c1 congenic strain containing a small NZB chromosome 4 genome on the B6 background, was identified as a likely candidate for the increase in B-1 cells.70 The lower expression of Cdkn2c was postulated to impair normal cell cycle arrest in B-1 but not B-2 cells because of inherent differences in their cell cycle regulation.
Two other loci already mentioned, Aem2 and Nba2, on chromosomes 7 and 1, respectively, were also confirmed in B6 congenic mice carrying the corresponding NZB locus.26,66 However, only low levels of anti-RBC and incomplete penetrance were observed in the Aem2 congenic mice, and for the Nba2 congenic mice, detection of anti-RBC required the addition of the lupus-enhancing Yaa (Y-linked accelerated autoimmunity) mutation (see section on BXSB mice for information about Yaa). Nba2 can also promote other lupus traits, such as IgG ANAs and severe GN in F1 hybrids of B6.Nba2 crosses with other lupus strains, such as the NZW.47 Further dissection of the Nba2 interval on chromosome 1 with a panel of subcongenic mice indicated at least two loci, one containing the SLAM (signaling lymphocyte activation molecule) family (see NZW genetics) and the other Fcγ receptors (FcγRs), that evidence now suggests additively contribute to autoimmunity.71,72 The subcongenic containing Ifi202, a candidate for Nba2, however, was not associated with lupus traits.43 Within the Fcγ interval, regulatory region variants of FcγRIIB that were associated with reduced expression were found in all major lupus-prone strains.73,74 This finding is supported by studies showing that deficiency of FcγRIIB promotes autoimmune susceptibility75 and further that the deficiency is related to the deficiency may result in to failure to adequately block IgG anti-DNA plasma cell generation.76
NZB mice are among the inbred strains of mice deficient in C5 owing to deletion of two base pairs (TA, positions 62-63) that lack terminal complement activity.77 Autoantibody-coated RBCs are therefore removed primarily by sequestration of agglutinated RBCs in the spleen and liver, through the use of Fc receptor–dependent phagocytosis, but not by complement-mediated hemolysis.78
New Zealand White Mice
The New Zealand white (NZW) mouse strain is of great interest because although it is clinically healthy, its genes can synergize with those of other lupus-prone and even normal strains to produce highly susceptible F1 hybrids or congenics.11,53,59,61,79–94 Therefore, the NZW genome likely contains controlling, repressor, or epistatic genes that protect from SLE, and such controlling genes must be powerful enough to allow the animal to effectively resist disease.
Clinical Characteristics and Autoantibodies
The NZW mouse has a slightly shortened life span and develops largely nonpathogenic autoantibodies, some of which are only intermittently detectable. The autoantibody pattern is characterized primarily by IgG antibodies to ssDNA and histones.86,95
Genetics
Lupus-affecting loci have been mapped to 12 chromosomes using crosses of NZW to lupus-predisposed and normal strains or by interval congenic mice containing introgressed NZW loci.53,86,96–101 Only a few have been further characterized, but they have provided significant insights. Of substantial interest because of its strong effect in human SLE is the MHC class II region, where NZW is H-2z and NZB is H-2d. Notably, BWF1 background mice expressing H-2d/z have a 30-fold greater risk of nephritis than H-2d/d mice.100 This increased susceptibility has been linked to class switching of various autoantibodies from IgM to IgG94 as well as antibodies to ssDNA, dsDNA, chromatin, and histones, but not to gp70.81–83 Also, within the MHC region is a variant NZW tumor necrosis factor alpha (TNF-α) gene with a polymorphism in the 3′UT region associated with lower TNF levels.84 Because of linkage disequilibrium within the MHC region, it has been difficult to separate the roles of class II and TNF; however, Fujimura and colleagues made three H-2 congenic BWF1 mice bearing distinct haplotypes at class II and TNF-α regions and showed that nephritis was affected by both the NZW MHC class II and the unique TNF-α allele.102 Also, within the MHC region is another recessive locus, Sles1 (Sle suppressor 1), which reduced the incidence of severe nephritis in B6-Sle1/Sle3 bicongenic and B6-Sle1/Yaa+ lupus-susceptible mice by about 50%.79,87,103 Thus, the MHC region of the NZW mice is rather complex with at least two lupus-promoting and one lupus-suppressing genetic variants.
Three other major NZW loci, Sle1, Sle2, and Sle3/5, on chromosomes 1, 4, and 7, respectively, have been studied in detail.103 They were derived from the mixed NZW/NZB background NZM2410 strain but are entirely NZW in origin except for an NZB region covering the telomeric third of Sle2. Sle1 is situated on the distal half of chromosome 1,85 overlapping with the NZB Nba2 locus, and contains many similar genetic variants. On the basis of panels of B6 subcongenic lines, Sle1 was subdivided into Sle1a, 1b, and 1c, and later these were further subdivided.103 Sle1a consists of two subloci, Slela.1 and Sle1a.2, with hyperactivity in B cells, ANA, and antichromatin tracking with Sle1a.1, whereas both Sle1a.1 and Sle1a.2 are required for hyperproliferation in CD4+ T cells and defects in CD4+ regulatory T (Treg) cells.86 Remarkably, the B6-Sle1a.1 subcongenic interval contains a single gene, Pbx1, a transcription factor in the TALE (three amino acid loop extension) family that participates in embryonic development and retinoic acid function.104 SPbx1 has no coding region variation; however, a higher level of a Pbx1-d isoform has been detected, which studies suggest promotes T-cell activation and inhibits retinoic acid–mediated apoptosis. Increased Pbx1 was also detected in T cells from patients with SLE.
Sle1b, which has the strongest effect on autoimmunity, promotes breaking of tolerance to chromatin.79,81 When NZW mice were compared with B6 mice, polymorphisms in the NZW (or NZM2410) involving at least 10 genes within the SLAM/CD2 gene cluster were identified,105 including expansion of 2B4 from one to four copies; coding changes in LY9, CD48, and CD84; transcription level variation in Ly108, CS1, CD84, and CD48; and differences in the predominant isoform of Ly108.106 Although dissecting the role of individual genes is difficult because of their close proximity, studies suggest that Ly108 is a major candidate that affects B- and T-cell tolerance and germinal center selection.107,108 The SLAM/CD2 family participates in a wide range of immune activities that encompass humoral immunity, cell survival, lymphocyte development, and cell adhesion,106,109 and it is likely that genetic variations promote autoimmunity by multiple mechanisms. The NZW Sle1b haplotype (haplotype 2) is present in most inbred and some wild strains, whereas the B6 haplotype 1 is found mainly in the C57BL-derived strains.106 Accordingly, B6 mice congenic for the Sle1b interval from normal haplotype 2 mice also develop lupus-like disease, further confirming the significance of this haplotype and providing evidence for susceptibility genes in the B6 strain.110
The most distal Sle1c is also associated with three subloci, two of which enhance autoimmunity in a chronic graft-versus-host disease (GVHD) model, and the third, which contains a variant complement receptor 2 (Cr2) gene, impairs humoral immune response and germinal center (GC) formation.111,112
The Sle2 locus on chromosome 4 promotes B-cell hyperactivity and B-1–cell expansion in B6.Sle2 congenic mice.69 It consists of at least three subloci, Sle2a and 2b of NZW origin and the aforementioned Sle2c from the NZB. The NZW-derived loci increase lymphocyte expansion and renal disease, whereas Sle2c, as mentioned previously, is associated with the increase in B-1 B cells.85 Sle2a also has been found to promote the loss of tolerance to DNA and alter splenic B-cell populations.113 Another locus, Sle3/5, is associated with generalized T-cell activation—elevated CD4:CD8 ratios, expansion of CD4+ T cells, and reduced apoptosis—caused by defective DCs and an intrinsic T-cell abnormality.114,115 Sle3/5, as its name implies, consists of two subloci, Sle3 and Sle5, that map to mid- and proximal chromosome 7, respectively.115 Single congenic B6.Sle3 or B6.Sle5 mice do not develop systemic autoimmunity, but double congenic mice that combine either of these with Sle1 develop splenomegaly, activated lymphocytes, ANAs, and glomerular immune complex deposits. The additive contribution of these lupus-predisposing loci to overall disease development has been further illustrated by the finding that single–Sle locus congenic mice had no to minimal autoimunity, but those with two Sle loci had intermediate severity that depended on the specific combination of loci, and triple congenic mice exhibited severe nephritis similar to that in the original NZM2410.80,87
In addition to the candidate genes associated with Sle loci, several other NZW variants that might modulate autoimmune susceptibility have been identified. The P2RX7 gene, within the Lbw3 region on chromosome 5, encodes the purinergic receptor P2X7, which initiates programmed cell death by aponecrosis, and NZW mice express the P2X-P allele associated with greater sensitivity to stimulation by adenosine triphosphate (ATP).62,89 It has been postulated that this variant might promote lupus by enhancing programmed cell death and increasing the release of cellular autoantigens such as nucleosomes. NZW mice also have a unique deletion of the T-cell receptor (TCR) α/β-chain gene, encompassing the Db2-Jb2 region on chromosome 6, which in one study was shown to segregate with lupus; however, this finding was not confirmed.57,58,92 In NZW mice, one of two murine Rt6 genes, Art2a-ps, on chromosome 7 is deleted.91 Rt6, a member of the family of mono–adenosine diphosphate (ADP)–ribosyl transferases, is a T cell-restricted, glycoprotein I (GPI)–anchored membrane protein that is activated by apoptosis and participates in DNA repair. It was suggested that the lack of Rt6 might increase susceptibility to lupus; however, no immune defects are associated with this variant, and an association with autoimmunity has yet to be established. Another NZW candidate susceptibility gene is CD22, a B-cell adhesion molecule that modulates BCR-mediated signal transduction, located between the Sle3/5 loci on chromosome 7.116 The NZW CD22a allele contains a 794-bp insertion in the second intron that causes altered splicing, the production of aberrant mRNA species, and reduced surface expression of the CD22 protein. Although it was not directly shown that this variant promotes lupus, heterozygous deficiency of CD22 was documented to markedly enhance the production of anti-DNA in Yaa+ mice.
(NZB/NZW) F1 Mice (BWF1)
The disease in BWF1 hybrid cross between NZB and NZW mice resembles human SLE in that disease is more severe and earlier in females, with high titers of IgG anti-dsDNA, antichromatin, ANAs, and LE cells occurring in virtually all; Treg- and B-cell networks fail, and death results from immune GN with tissue damage (Tables 17-1 to 17-3 and Box 17-2).10,26 Involvement of the innate immune system, elevation of BAFF, and increased type-1 IFN–induced gene expression are similar to features in human lupus.117–133 Both NZB and NZW parents contribute genetically to the immune abnormalities that cause disease, as discussed in the preceding sections. The B-cell hyperactivity characteristic of the NZB is inherited by the BWF1, with abnormally high secretion of Ig being detectable by 1 month of age.11 However, the T-cell dependence of the response is more striking than in the NZB parent and is responsible for the isotype shift from IgM anti-DNA to IgG anti-DNA that precedes clinical disease.134
Box 17-2
Characteristics of (NZB/NZW) F1 Mice
Histologic
1. Glomerulonephritis with proliferative changes in mesangial and endothelial cells of glomeruli, capillary basement membrane thickening, and chronic obliterative changes; mononuclear cell infiltrates in interstitium.
2. Glomerular immune deposits of IgG (predominantly IgG2a) and C3; similar deposits in tubular basement membrane and interstitium.
3. Thymic cortical atrophy by 6 months of age, including epithelial cell atrophy.
4. Myocardial infarcts with hyaline thickening of small arteries.
Autoantibodies
1. IgG anti-dsDNA (also binds single-stranded DNA), enriched in IgG2a and 2b.
2. Antinuclear antibody (ANA) and lupus erythematosus (LE) cells in all.
3. IgG antibodies bind chromatin, nucleosomes, and phospholipids.
4. Antithymocyte in most females and some males.
5. Renal eluates contain IgG anti-dsDNA concentrated 25 to 30 times greater than in serum; IgG2a isotype is dominant.
6. Modest elevations of circulating immune complexes; these include glycoprotein (gp) 70–anti-gp70.
7. Low serum complement levels by 6 months of age in females.
Immune Abnormalities
1. Polyclonal B-cell activation.
2. B cells are resistant to tolerance to some antigens.
3. Strict dependence on T-cell help for formation of pathogenic IgG anti-DNA, CD4+CD8−, and CD4−CD8− α/β TCR cells, as well as CD4−CD8− γ/δ TCR cells, can provide help.
4. IgG repertoire becomes restricted with age to certain public idiotypes; there is some restriction of B-cell clonality in the IgG anti-DNA response.
5. Clearance of immune complexes by Fc- and complement-mediated mechanisms is defective.
6. Disease and autoantibody production are sensitive to sex hormone influences.
Genetics
1. The expression of high-titer IgG anti-dsDNA requires heterozygosity at the major histocompatibility complex, namely, H-2(d/z).
2. Additional complementary non–H-2-linked genes are required from both NZB and NZW parents to permit full expression of the IgG anti-DNA response. By microsatellite analysis of DNA, there are approximately 10 genes on as many chromosomes, with multiple genes required for early mortality, glomerulonephritis, antichromatin, and splenomegaly; this suggests a multigenic inheritance, with certain groupings predisposing more strongly than others to disease, rather than a simple additive model. NZW also provides a resistance gene.
3. The large deletion in the β chain of the TCR of the NZW parent probably does not predispose to disease.
dsDNA, double-stranded DNA; Ig, immunoglobulin; TCR, T-cell receptor.
Clinical Characteristics and Autoantibodies
Large quantities of IgG antibodies that bind nucleosomes, chromatin, dsDNA, and ssDNA in BWF1 mice are striking and can be abrogated by removal or inactivation of CD4+ T cells.94,135 IgG antibodies to dsDNA contain subsets that cause nephritis. Transfer of selected monoclonal BWF1 IgG2 anti-dsDNA antibodies to normal BALB/c mice induces nephritis.136,137 Infusion of anti-DNA into rodent kidneys induces proteinuria,138 and normal mice secreting BWF1 IgG anti-dsDNA (encoded by transgenes) develop GN.139 Anti-DNA, antinucleosome antibody, and immune complexes containing gp70 and anti-gp70s all contribute to nephritis.140,141 ANAs are detectable in most 3-month-old females; they include antibodies that bind subnucleosomes, nucleosomes, chromatin, dsDNA, ssDNA, dsRNA, transfer RNA (tRNA), polynucleotides, and histones.134–141 IgM anti-DNAs arise first; by 6 months of age, IgG anti-DNAs appear.10,134 The IgG 2a and 2b subclasses are most common; these subclasses fix complement and bind FcγRs. Antibodies to erythrocytes are found in 35% to 78% of BWF1 females but rarely cause hemolytic anemia. Approximately 50% of females develop NTAs by 6 months of age. Because the genes governing NTA, anti-DNA, and antierythrocyte antibodies probably segregate separately,* New Zealand mouse strains have been bred that have high-titer NTAs but no autoimmune disease. However, NTAs, by altering T cell functions, may serve as accelerators of the disease process that occurs in mice with IgG anti-DNA. Both IgM and IgG antiphospholipid antibodies have been detected and obtained as monoclonal antibodies from BWF1 mice.150 Some have anticardiolipin (aCL) activity, and others lupus anticoagulant properties. However, clotting disorders are not characteristic of BWF1 mice. Antibodies to ubiquitin and fibrillarin have been reported, as have cryoglobulins.151–153
Nephritis in BWF1 Females: the Autoantibodies, the Infiltrating Cells, and the Predisposing Glomerular Structures
Shortly after the switch from IgM to IgG, IgG and complement deposit in the mesangia of BWF1 glomeruli, spreading later to capillary loops and interstitial tubular regions.10 Proteinuria appears between 5 and 7 months of age; azotemia followed by death occurs in females at 6 to 12 months of age. Approximately half of females are dead by 8.5 months and 90% at 12 months.10,136
Antibodies eluted from BWF1 glomeruli are composed predominantly of IgG anti-DNA/antinucleosome antibody (anti-NUC); 50% of the total IgG is anti-DNA according to some reports.154,155 In our laboratory, anti-DNA accounts for as much as 85% of the total glomerular IgG.156 IgG2a is the dominant isotype in glomerular deposits, suggesting a role for Th1 cells, because production of IgG2a depends on IFN-γ. Immune complexes containing gp70/anti-gp70 are also found in glomeruli of BWF1 mice. Gp70 is an endogenous retroviral glycoprotein produced by hepatic cells that is found in all mouse strains but is quantitatively higher in some, including the main lupus-prone strains.157 Among the IgG anti-dsDNA antibodies made by BWF1 mice are subsets that cause nephritis by (1) passive trapping of immune complexes containing them, (2) direct attachment to planted (e.g., chromatin/nucleosomes/DNA) or cross-reactive (e.g., laminin) glomerular and tubular antigens, and (3) cationic charge, which binds to anionic glomerular areas.141,156 For gp70/anti-gp70 immune complexes, passive trapping is probably the major mechanism. NZB chromosome regions Nba2 (on chromosome 1; contains the Ifi202 gene) and H2 (on chromosome 17) are linked both to high levels of gp70/anti-gp70 immune complex production and to high titers of IgG anti-DNA.142 Plasma cells making antibodies to dsDNA and to histone 2B are found in renal tissue of BWF1 mice as well as in their bone marrow and spleens,158,159 so local synthesis of nephritogenic antibodies occurs. Regarding the role of anti-dsDNA/NUC in BWF1 nephritis, the availability of apoptotic chromatin planted in glomerular membrane is important in pathogenicity.160 Furthermore, BWF1 mice have lower levels of renal DNAse1 after the development of nephritis; thus nucleosomal DNA fragmentation during apoptosis is decreased in kidney tissue, and partial fragmentation may induce more production and binding of anti-DNA/NUC.161 In support of this idea, one study has reported that administration of heparin to BWF1 mice increased enzymatic degradation of nucleosomes, inhibited their binding to laminin and collagen, reduced glomerular deposition of IgG, and delayed development of nephritis.162 Various epitopes in chromatin can be targeted by autoantibodies: some antibodies against apoptotic chromatin recognize acetylated epitopes.163 Epigenetic changes in DNA, including acetylation and methylation, are controlled in part by microRNA; at least three dysregulated miRNAs have been found in splenocytes of BWF1 mice at the onset of nephritis.164 Finally, some BWF1 antibodies to DNA/NUC can bind to and/or penetrate living cells; some induce proliferation of glomerular cells and impair intracellular production of protein.165–167
Other antigens and antibodies have been reported in glomerular eluates, including antihistones, anti-C1q, and anti-RNA polymerase.168,169 Hypocomplementemia occurs concomitantly with high serum levels of IgG anti-DNA.10
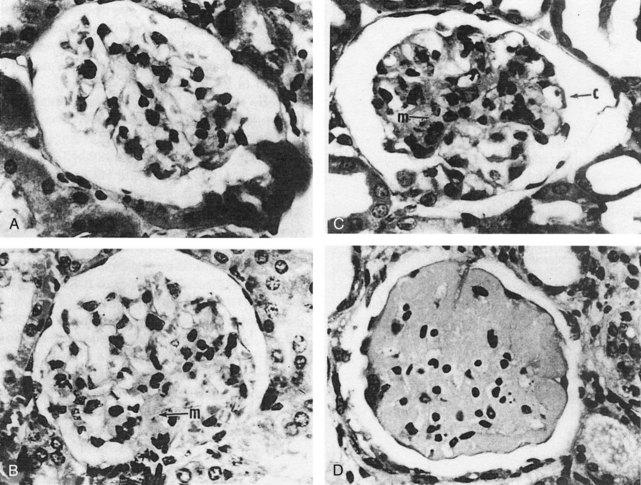
Histologic changes in kidneys include chronic obliterative changes in glomeruli, mesangial and peripheral proliferative changes, capillary membrane thickening, glomerular sclerosis, tubular atrophy, infiltration by mononuclear lymphocytes and monocyte/macrophages, and vasculopathy (primarily degenerative, occasionally inflammatory) (Figure 17-1).10,26 Some studies suggest that after deposition of complement-fixing IgG in glomeruli, the next local abnormality is upregulation of genes associated with macrophage activation,120,170 followed later by upregulation of genes characteristic of T and B cells. Successful treatment of nephritis is associated with downregulation of the initial macrophage signature.118,120 These tissue macrophages derive from peripheral blood GR1lo monocytes.117 At the beginning of nephritis, the renal F4/80hiCd11cint macrophages upregulate cell surface CD11b, acquire cathepsin and matrix metalloproteinase activity, accumulate autophagocytic vacuoles, and upregulate expression of proinflammatory and tissue repair/degradation genes.171 In contrast, dendritic cells (F4/80loCD11chi) appear in kidneys later, after proteinuria onset, and disappear more rapidly after treatment.117 Renal infiltration of T and B lymphocytes that follows glomerular/tubular innate cell infiltration is associated with upregulation of MHC class II on T cells and secretion of IFN-γ.120 Inhibition of macrophage migration inhibitory factor (which promotes retention of monocytes in tissue via the CD74 receptor) reduces leukocyte accumulation and expression of proinflammatory cytokines and chemokines in renal tissue of BWF1 mice.172 Additional evidence for the crucial role of monocytes/macrophages in BWF1 nephritis are the observations that renal inflammation after Ig deposition is abrogated by knocking out activating gamma globulin FcRs in infiltrating monocytes/macrophages but not by impairing FcRs on mesangial cells.170 Apart from infiltrating inflammatory cells, glomeruli in BWF1 mice differ from nonautoimmune mice in ways that may promote glomerular disease. Embryonic forms of collagen IVα chains are more abundant than in normal strains,173 and IL-20 (in the IL-10 family) and C-IVα receptors are upregulated in mesangial cells. Activation of these receptors upregulates expression of the chemokines/receptors monocyte chemoattractant protein-1 (MCP-1) and RANTES (regulated upon activation, normal T-cell expressed, and secreted) as well as mediators of oxidative damage inducible nitric oxide synthase (iNOS) and reactive oxygen species (ROS).174 BWF1 mice are also prone to chronic nephritis because their renal tissue expresses high levels of the inflammatory chemokine CXCL13/BCL, which attracts leukocytes into interstitial areas.172 Upregulation of the canonical Wnt/beta-catenin pathway is also a characteristic in BWF1 renal tissue as nephritis develops, paralleled in renal tissue and serum by increase in the Wnt inhibitor Dkk-1.175 Dkk-1 at these high levels can induce apoptosis in mesangial and renal tubular cells—an additional factor contributing to tissue damage. In summary, a combination of multiple hematopoietic cells attracted into renal tissue after deposition of autoreactive Ig and activation of complement mediate acute and chronic renal disease in BWF1 mice, and the renal tissue itself differs from that of non-lupus strains in ways that make the tissue more susceptible to damage.
Neurologic Tissue
IgG1 antibodies have been eluted from the neurons of BWF1 mice176; it is not known whether they cross-react extensively with lymphocytes, as do some human lupus antineuronal antibodies. BWF1 mice exhibit anxiety-like behavior and have inflammatory infiltrates and deposition of IgG and C3 in hippocampi.177
Lymphoproliferation
The lymphoproliferative features of NZB mice occur in BWF1 hybrids, which exhibit mild lymphadenopathy and splenomegaly.10 Lymphoid neoplasia is far less common in BWF1 than in NZB mice. Some investigators have reported a relatively high incidence of thymoma, from 1% to 5%,178 but that has been rare in our colonies unless mice are treated with cytotoxic agents.179 Extrarenal lesions occur in BWF1 mice, including lymphocytic infiltration of salivary glands, mild inflammation around bile ducts in the liver, pancarditis, vasculitis (less common than in MRL-Fas[lpr] and BXSB mice), myocardial infarcts, and deposits of DNA and anti-DNA in the dermoepidermal junction of skin and in the choroid plexus.10,180
Sex Hormone Influences on Lupus in BWF1 Mice
Female BWF1 mice have earlier and more severe autoimmune disease than males. Most BWF1 males develop ANAs, including antibodies to DNA, but the switch from IgM to IgG occurs late in life, usually after 12 months. Histologic evidence of nephritis can be found in males, and most die of slowly progressive chronic nephritis by 15 to 20 months of age.10
The BWF1 mouse is particularly sensitive to the effects of sex hormones on disease. In general, androgens are protective and suppress the expression of autoantibodies and disease, and estrogens are permissive.181–186 Males that are castrated and/or treated with estrogens or testosterone antagonists assume a female pattern: early IgM-to-IgG switch of anti-DNA antibodies and early, fatal nephritis. Females that are treated with castration and androgens, or antiestrogens, have prolonged survival, with suppression of IgG anti-DNA and nephritis.181,185,187 Administration of ethinylestradiol to BWF1 mice accelerates disease.182 In old females, androgens can suppress disease without altering the elevations of IgG anti-DNA.181 Female and male BWF1 mice deficient for estrogen receptor alpha (ERα) have lower blood levels of IgG anti-dsDNA and IFN-γ than ERα-intact controls, and increased survival.185 In addition, expression of several sex hormone–regulated genes in antigen-presenting splenocytes is different in female and male BWF1 mice.188 Prolactin also influences BWF1 disease: administration of prolactin accelerates disease, whereas bromocriptine suppresses it.189–191 Continuous treatment of premorbid BWF1 females with high doses of depot medroxyprogesterone acetate reduces IgG2a deposition in glomeruli, histologic glomerular damage, proteinuria, and mortality.192 These benefits may relate to the ability of progesterone to suppress activation of plasmacytoid DCs (pDCs) and their production of IFN-α, whereas 7beta-estradiol (E2) can increase pDC activation and cytokine production, depending on the age of the BWF1 mouse at the time of treatment.193
There are receptors for estrogens, progestogens, and prolactin on lymphocytes and natural killer (NK) cells.191,194 The administration of estradiol in vivo dramatically suppresses NK cell function, and NK cells downregulate activated B cells.194 In addition, normal mice transgenic for a murine IgG antibody to DNA show defective B-cell tolerance if they are treated with exogenous estradiol.195 Such mice fail to delete B cells that are producing anti-DNA from unmutated germline genes; this failure of negative selection is particularly marked at the transitional cell type 1/type 2 selection checkpoint.196 Data now suggest that estradiol is linked to SLE in part via effects on type 1 IFN pathways. For example, female BWF1 mice have higher expression of Irf5 mRNA (Irf5 is a molecule in the pathway of plasmacytoid dendritic cell activation that results in type 1 IFN production) than both BWF1 males and normal mice; and those levels are reduced in ERα−/− mice.131 Activation of spleen cells by IFN (either α or γ) upregulates expression of ERα, and both IFN-α– and ERα–responsive genes are upregulated in BWF1 females.128,197 In vivo treatment of castrated BWF1 males with 17β-estradiol increases steady-state levels of Ifi202 mRNA in splenic cells (Ifi202 is an interferon-inducible gene); dihydrotestosterone decreases those levels. In contrast, Ifi202 mRNA levels are not detectable in female BWF1 mice that were Esrα−/−. Thus, female and male hormones differentially regulate the expression of some IFN-inducible genes, including Ifi202, which has been suggested to be a susceptibility gene for murine lupus.198
Interferons and SLE in BWF1 Mice
High levels of IFN-γ in plasma and lymphoid tissues are characteristic of BWF1 mice. IFN-γ is a major cytokine produced by Th1 cells, which provide help to B cells. The high IFN-γ levels may relate to low levels of the negative regulator suppressor of cytokine signaling 1 (SOCS-1).199 Elevated genetic signatures associated with type 1 IFN (e.g., IFN-α) are also characteristic of BWF1 mice, similar to those in human SLE.133 Enhanced expression of IFN-α (by infection of their cells with DNA encoding IFN-α) increases short-lived plasma cells in healthy and BWF1 mice, but only the BWF1 mice make autoantibodies and develop accelerated lupus129; this acceleration requires CD4+ T-cell help.130 Conversely, inhibition of IFN-α by inhibitory antibodies induced by vaccination with an IFN-α kinoid protect mice from severe nephritis.132
Abnormalities of Hematopoietic Cells in BWF1 Mice
BWF1 mice exhibit the hyperactivated B-cell phenotype of their NZB parent, except that defects appear later with abnormally elevated IgM occurring by 1 month of age. Pre-B lineage cells can partially transfer disease: severe combined immunodeficiency disease (SCID) mice (a mutant strain that lacks most T cells) inoculated with BWF1 bone marrow pre-B cells develop autoantibodies (including IgG anti-dsDNA) and, in approximately 25%, clinical nephritis.200 Thus, BWF1 B cells alone can induce lupus-like disease, albeit less globally than in the presence of T-cell help; the B-cell repertoire that expresses anti-DNA is also somewhat restricted. In this regard, public idiotypes (Ids) that are expressed on total serum IgG become increasingly restricted as the mice age.201 Although many different V genes can be used to assemble antibodies that bind DNA,202 most BWF1 anti-DNA monoclonal antibodies belong to one of approximately 12 families.203–205 This type of restriction is seen in normal, antigen-driven antibody responses. In BWF1 mice, B-1 B cells and MZ B cells are increased in number. Depleting some of these cells by administering a B-cell superantigen (protein A from Staphylococcus aureus) delays appearance of serum IgG anti-DNA and reduces proteinuria, further suggesting participation of these cells in autoimmunity in this strain.206 Depleting mature B2 cells (MZ) plus peritoneal B1 cells can be achieved by repeated doses of anti-mouse CD20 plus a BR3-Fc fusion protein (which blocks BAFF): These treatments delay disease in young mice and prolong life in old nephritic mice without reducing autoantibody levels.207 Murine B cells from spleen, bone marrow, and peritoneum can express BAFF when activated via innate immune pathways; in BWF1 mice, splenic MZ and germinal center B-cell populations express high levels of BAFF,127 indicating their continued activation.
Abnormalities of Thymus and T Cells
The characteristic degeneration of thymic epithelial cells seen in NZB mice at 1 month of age also occurs in BWF1 mice, but at 6 months.11 Responses to thymectomy have been variable; there are reports of thymectomy failing to alter disease or even accelerating it.46 Full-blown BWF1 lupus depends on the presence of CD4+ Th cells; T-cell lines from nephritic mice can accelerate disease in naive young syngeneic mice.208,209 Elimination or inactivation of CD4+ T cells prevents the onset of disease and can even partially reverse established nephritis.135,210 As BWF1 mice age, the numbers of CD4+ T cells increase fivefold, and these cells are polyclonal.211,212 T cells from nephritic BWF1 mice can drive B cells from young normal mice to make pathogenic autoantibodies,208,209 but T cells from young mice do not have this property. Several T-cell subsets influence BWF1 disease. These include Th1 cells (require IL-12 for development; secrete IFN-γ; mediate cell-mediated immunity; express Tbet transcription factor), Th2 cells (require IL-4 for development; support B-cell production of autoantibodies; express transcription factor GATA3), Th17 cells (require transforming growth factor beta [TGF-β] plus IL-6 plus IL-2 for development; secrete proinflammatory IL-17, which attracts neutrophils; require IL-21 for maintenance; help B cells make autoantibodies; express the protein RORγT), and Treg cells (CD4+CD25+; require TGF-β plus IL-2 for development; may secrete TGF-β or IL-10; downregulate CD4+ Th cells and autoreactive B cells by contact; express the protein FoxP3). Another major Th cell promoting IgG autoantibody formation is located in lymphoid follicles: these follicular CD4+ Th (TFH) cells stimulate GC B cells to produce autoantibodies via interactions between inducible T-cell co-stimulator) ICOS and its ligand, B7-related protein 1 (B7RP-1). Interrupting second signals between these T cells and B cells alters BWF1 disease.212,213 In addition, blockade of the CD28/cytotoxic T-lymphocyte antigen 4 (CTLA-4) T-cell surface molecule’s interactions with CD80/CD86 (also called B7-1/B7-2) on APCs prevents disease. Experiments showing this include the administration of CTLA-4–Ig, which binds to CD80 and CD86, thus preventing interaction with CD28,214 and the administration of antibodies to CD80 and CD86.215 In addition, blocking second signals that activate B cells (CD40 interacting with CD40 ligand [CD40L]) by administration of antibody to CD40L prolongs survival in BWF1 and other New Zealand–background lupus mice.216,217 Blocking both CD28/B7 and CD40/CD40L interactions is probably more effective than blocking either one alone.218 This multiple-blockade strategy was used in one study to reverse nephritis in BWF1 mice, along with doses of cyclophosphamide, but disease recurred when the therapies were stopped.120 Enhancing proinflammatory T-cell function by administration of IL-6 accelerates disease, and antibodies to IL-6 delay it.219 Administration of anti–IL-10, which also suppresses IL-6, delays disease in BWF1 mice.220
TGF-β is essential for the suppression by some CD4+CD25+ Treg cells221 and by CD8+ T cells,222 which delay autoimmunity in BWF1 mice tolerized with histone or Ig peptides; a similar process protects healthy mice from autoimmunity.223 However, late in disease TGF-β contributes to glomerular scarring and thus to shortened survival.1 IL-1 and TNF-α are both proinflammatory and may be abnormally elevated in BWF1 lupus.224 The role of TNF-α in murine and human lupus has been debated for several years. In support of its role, NZW mice have a variant gene that encodes lower levels of TNF-α, and short-term administration of TNF-α to BWF1 mice delays disease.225 However, long-term administration of the cytokine worsens disease.226
Abnormalities of Monocytes/Macrophages
Monocytes/macrophages are primary sources of IL-1, production of which is reduced in BWF1 and other murine lupus strains.227 Macrophages also produce IL-12, the major cytokine stimulating Th1 responses. The ability of C-reactive protein (CRP) treatment to delay disease onset in BWF1 mice may relate to the fact that CRP reduces IL-12 production by macrophages following ingestion of apoptotic materials; those macrophages have reduced ability to activate T cells.228–231 Monocytes that differentiate into macrophages in renal tissue (and possibly others that express activating FcR) seem to govern the inflammatory response to Ig deposition and complement activation in BWF1 kidneys; those cells are discussed in the previous section on nephritis.120,170 BWF1 monocytes and dendritic cells exposed to apoptotic cells can activate Th cells in BWF1 mice, consistent with the presentation of autoantigens by APCs, but the T-cell responses are more vigorous in lupus-prone mice than in normal mice.232
The Role of Defective Regulatory Cells in BWF1 Lupus (CD4+CD25+, CD8+, NK T Cells, B-1 B Cells)
Finally, there is strong evidence that numbers and functions of regulatory cells that ordinarily suppress activated T and/or B cells are defective in BWF1 F1 mice. As the mice age, CD8+ cytotoxic/suppressive T cells fail to expand, whereas CD4+ T and B cells are increasing greatly in numbers, and very few CD8+ cells express surface markers of activation and memory. Furthermore, stimulation of CD8+ T cells from old BWF1 mice results in apoptosis rather than activation.212 Tolerizing regimens with autoantibody- or histone-derived peptides induce both suppressive CD8+ T cells and classic CD4+CD25+ Treg cells, each of which can prolong survival in BWF1 or (NZB/SWR) F1 mice, indicating that Treg-cell defects can be “repaired” in vivo.221–233 The regulatory capacity of CD8+ T cells depends on their expression of Foxp3 and of programmed death 1 (PD-1), a member of the CTLA-4 family that helps determine whether a CD8+ T cell has suppressive capabilities.234 Regulatory B cells have also been described; these cells (called B-10 cells) in mice are CD1dhiCD5+ B cells (B-1 cells) that secrete IL-10. B-10 B cells expand as disease develops in BWF1 but are not able to overcome the effects of hyperactivated autoantibody-producing plasma cells.235 CD1-restricted NK T cells prevent the development of autoimmune manifestations if activated in early stages of disease in BWF1 (nephritis), pristane-injected BALB/c (nephritis), and MRL-lpr (dermatitis) mice,1,4 but not in late stages of disease in BWF1 or pristane-injected SJL mice.236,237 As BWF1 mice age, NK T cells expand in number and become hyperactive; they can actually increase production of IFN-γ—a major cytokine that, as noted previously, enhances SLE in this strain.238
Abnormalities of Dendritic Cells in BWF1 Mice
DCs, which connect innate and acquired immunity, can be activated by RNA and/or DNA produced by viruses and bacteria and by patients with SLE, in complex with antibodies to nucleoproteins. As BWF1 mice age, DCs expand in number and acquire the ability to attract B cells and to present antigen.239 This activity is particularly brisk in the spleen, where DCs stimulate nucleosome-reactive T cells to a much greater extent than normal,240 promoting induction of autoantibodies to apoptotic materials.241 Furthermore, pDCs are a major source of type 1 IFNs. High production of these IFNs is characteristic of BWF1 mice and of humans with SLE.6 Deficiency of the type I IFN receptor protects NZB mice from disease,242 and administration of IFN-α accelerates it.243 Therefore, abnormal DCs and their production of IFN-α play a critical role in promoting lupus-like disease in BWF1 mice.
Genetic Predisposition
Genetic predisposition is reviewed in the preceding sections on NZB and NZW mice. In BWF1 mice, genetic contributions to disease are provided by both NZB and NZW parents. Although in F1 hybrids these must be dominantly transmitted, most loci identified in mapping and congenic studies of NZB and NZW loci exhibit additive inheritance. The most important contributors are the MHC genes (heterozygosity for H-2 d/z) and loci on chromosome 4 (Lbw2, Nba1) that were shown to directly affect autoimmunity in BWF1 mice.6,43 Likely of major importance are genes on chromosome 1 from the NZB (Nba2) and NZW (Sle1) and chromosome 7 from the NZW (Sle3/5). In addition, multiple non-MHC genes on at least eight different chromosomes contribute to disease susceptibility.56–72,74–81,83–88
(SWR × NZB) F1 (SNF1) Mice
The SNF1 mouse is a model of lupus nephritis that is produced by mating the normal Swiss Webster (SWR) mouse with the autoimmune NZB mouse (Box 17-3).244–246 In contrast to NZW mice that mated with NZB to produce the BWF1 strain, SWR mice are completely healthy, with normal life spans, low levels of serum gp70, and no evidence of autoimmune disease.244 Their B cells can produce Igs bearing the same public Ids that dominate serum Ig in MRL-Fas(lpr) mice.247,248
Box 17-3
Characteristics of NZB X SWR F1 (SNF1) Mice
Immune Abnormalities
1. B cells are hyperactivated.
2. The development of nephritis depends on the presence of T-cell help for production of IgG anti-DNA.
3. Cationic IgG anti-dsDNA may use the allotype of either the NZB or healthy SWR parent.
4. Anti-dsDNA deposited in glomeruli cluster into two main groups defined by their idiotypes.
5. CD4+D8− and CD4−CD8− T cells can provide help for the synthesis of cationic IgG anti-dsDNA.
Related posts:
 The Innate Immune System in SLE
Clinical Aspects of the Antiphospholipid Syndrome
Gastrointestinal and Hepatic Manifestations
Novel Therapies for SLE: Biological Agents Available in Practice Today
Mixed Connective Tissue Disease and Undifferentiated Connective Tissue Disease
Pathogenetic Mechanisms in Lupus Nephritis
The Innate Immune System in SLE
Clinical Aspects of the Antiphospholipid Syndrome
Gastrointestinal and Hepatic Manifestations
Novel Therapies for SLE: Biological Agents Available in Practice Today
Mixed Connective Tissue Disease and Undifferentiated Connective Tissue Disease
Pathogenetic Mechanisms in Lupus Nephritis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


