Adult Motor Neuron Disease*
Lisa S. Krivickas
Gregory T. Carter
INTRODUCTION
Adult motor neuron disease (MND) is often considered synonymous with amyotrophic lateral sclerosis (ALS). In the United States, the term ALS is frequently used to describe all forms of adult-onset MND. However, it is also used to refer specifically to the most common form of adult MND, which is sporadic, acquired ALS. In the United Kingdom, the converse is true where the generic term for all forms of ALS is MND (1). In reality, adult MND actually encompasses a group of disorders that include ALS; primary lateral sclerosis (PLS); progressive muscular atrophy (PMA); progressive bulbar palsy (PBP); adult-onset, progressive spinal muscular atrophy (SMA); and X-linked, recessive spinobulbar muscular atrophy (SBMA). A presentation with pure upper motor neuron (UMN) signs may be called PLS, whereas pure bulbar presentation may be called PBP, and pure lower motor neuron (LMN) presentation called PMA. Whether these conditions exist as distinct diseases or rather represent part of the spectrum of ALS is still debated. This is represented schematically in Figure 28-1.
At least one form of PLS with a benign course and autosomal dominant inheritance has been reported (2, 3). ALS is also referred to as Lou Gehrig’s disease, named after perhaps the most famous person yet afflicted with the disease. SBMA is commonly referred to as Kennedy’s disease, named after the physician who first described this disorder in 1968 (4).
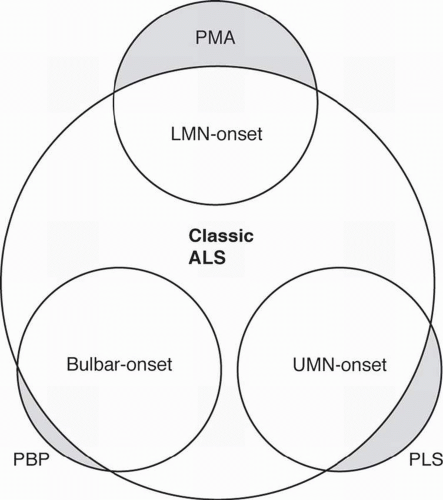 FIGURE 28-1. Schematic representation of adult MNDs. |
For the purposes of this chapter, we will use the terms ALS, PLS, PMA, PLS, SMA, and SBMA. In the adult population, ALS is far more common than the other disorders. Thus, ALS will constitute most of the focus of this chapter, which starts first with a description of the diseases, including epidemiology and genetics. This is followed by diagnostic workup, including electrodiagnosis, pharmacological management, and rehabilitation strategies, most of which may be applied to any of the adult MNDs.
OVERVIEW OF THE MAJOR ADULT MNDs
Amyotrophic Lateral Sclerosis
ALS is a rapidly progressive neuromuscular disease that destroys both UMNs and LMNs, resulting in spasticity and diffuse muscular atrophy and weakness. The vast majority of ALS cases are presumably acquired and occur sporadically. However, approximately 10% of all ALS cases are familial amyotrophic lateral sclerosis (FALS) and usually inherited as an autosomal dominant trait. About 15% of these cases result from a gene defect on chromosome 21q12.1, which leads to a toxic gain of function in the antioxidant enzyme Cu/Zn superoxide dismutase (SOD1) (5, 6). Over 100 unique SOD1 mutations have been identified (5, 6, 7). Emerging evidence suggests that these mutations result in increased oxidative stress for the motor neurons, leading to cell death, which is felt to be related to free radical toxicity (6, 7).
The etiology of sporadic amyotrophic lateral sclerosis (SALS) and the other 85% of FALS is unknown. Increasing data suggest that excessive glutamate activity in the brain and spinal cord may play an important role. Glutamate is one of the main central nervous system (CNS) excitatory neurotransmitters in the brain, and excess levels of this chemical have been demonstrated in the serum, spinal fluid, and brain tissue of ALS patients (8, 9). There appears to be reduced clearance of glutamate from critical motor control areas in ALS as well as decreased levels of glutamate transport protein (10, 11).
Epidemiology of ALS
ALS most commonly strikes people between 40 and 60 years of age with a mean age of onset of 58 years (12, 13, 14). The overall
prevalence rate in the worldwide population is somewhere between 5 and 7 per 100,000, making it one of the most common neuromuscular diseases worldwide (15). Further, population studies suggest that the incidence of ALS is increasing, although this is probably due, in large part, to people living longer and better recognition of the diagnosis (16, 17). There appears to be a higher incidence in urban areas, felt to be related to environmental factors (17, 18, 19). The association of nutrient intake with the risk of ALS was investigated in a population-based case-control study conducted in three counties of western Washington State from 1990 to 1994 (20, 21). The authors found that alcohol consumption was not associated with increased risk of ALS. Ever having smoked cigarettes was associated with a twofold increase in risk and a greater than threefold increased risk was observed for current smokers. Further, dietary fat intake was associated with an increased risk of ALS, while dietary fiber intake was associated with a decreased risk. Interestingly, consumption of antioxidant vitamins from diet or supplement sources did not alter the risk but glutamate intake was associated with an increased risk of ALS. The finding that cigarette smoking and glutamate consumption are risk factors for ALS is consistent with current etiologic theories that implicate glutamate excitotoxicity and oxidative stress in the pathogenesis of ALS. The associations with fat and fiber intake warrant further study and biologic explanation.
prevalence rate in the worldwide population is somewhere between 5 and 7 per 100,000, making it one of the most common neuromuscular diseases worldwide (15). Further, population studies suggest that the incidence of ALS is increasing, although this is probably due, in large part, to people living longer and better recognition of the diagnosis (16, 17). There appears to be a higher incidence in urban areas, felt to be related to environmental factors (17, 18, 19). The association of nutrient intake with the risk of ALS was investigated in a population-based case-control study conducted in three counties of western Washington State from 1990 to 1994 (20, 21). The authors found that alcohol consumption was not associated with increased risk of ALS. Ever having smoked cigarettes was associated with a twofold increase in risk and a greater than threefold increased risk was observed for current smokers. Further, dietary fat intake was associated with an increased risk of ALS, while dietary fiber intake was associated with a decreased risk. Interestingly, consumption of antioxidant vitamins from diet or supplement sources did not alter the risk but glutamate intake was associated with an increased risk of ALS. The finding that cigarette smoking and glutamate consumption are risk factors for ALS is consistent with current etiologic theories that implicate glutamate excitotoxicity and oxidative stress in the pathogenesis of ALS. The associations with fat and fiber intake warrant further study and biologic explanation.
Considerable clustering has been demonstrated in the Western Pacific region of the world (15, 16, 17). Other sporadic cluster cases have been reported but without obvious environmental or causal factors (15). Men appear to be more commonly affected than women with a male-to-female ratio of about 1.5:1.0 (12).
Poor prognostic factors include older age at time of onset, bulbar and/or pulmonary dysfunction early in the clinical course of the disease, short time period from symptom onset to diagnosis, and predominance of LMN findings at the time of diagnosis (12, 14, 15, 16).
More women than men present with bulbar symptoms, and the progression of bulbar palsy appears to be more rapid in women (20, 21). Young males with ALS may have a longer life expectancy but overall the median 50% survival rate is 2.5 years postdiagnosis, except in patients with primary bulbar symptoms, where the 50% survival rate is only 1 year (22). Survival rates will obviously vary to a degree depending on the patient’s decision to use or not use mechanical ventilation and a feeding tube (23). Nonetheless, by 5 years postdiagnosis the overall survival rate is only 28% (12, 14, 15).
Atypical, “ALS-like,” MNDs have been reported infrequently as a remote complication of several malignancies, including lymphoma and small cell carcinoma of the lung (24, 25). These likely represent paraneoplastic syndromes and not a true manifestation of ALS (26). Regardless, patients with atypical MND should be screened for malignancy.
Spinal and Spinobulbar Muscular Atrophy
There are many forms of SMA, all of which involve selective destruction of anterior horn cells. The various forms of SMA are clinically dissimilar, with some rare forms affecting distal or bulbar muscles only. The most common forms are often referred to as types I, II, and III (27). These are mostly disorders of childhood and are usually inherited as autosomal recessive traits. SMA I, also known as Werdnig-Hoffman disease (WHD) or acute, infantile-onset SMA, is a severe disorder resulting in death before age 2 years. SMA II, also referred to as early-onset, intermediate SMA or chronic WHD, is less severe, with signs and symptoms becoming apparent in the first 6 to 18 months of life. SMA III, also known as Kugelberg-Welander disease (KWD), is a chronic, later onset disorder, associated with significantly less morbidity. Signs and symptoms of SMA III usually become apparent between ages 5 and 15 years. In prior studies looking at SMA II and III over a 10 year period, SMA II subjects showed marked weakness and progressive decline of strength while SMA III subjects had a relatively static or very slowly progressive course and were far stronger. In both SMA II and SMA III, proximal weakness was greater than distal. Joint contractures, progressive scoliosis, and restrictive lung disease (RLD) were present in most of the SMA II individuals but these complications were rare in SMA III (27). Scoliosis in a patient with SMA II is shown in Figure 28-2.
There are two forms of SMA that have onset in the adult age group. One is an adult-onset form of SMA with age of onset of 17 to 55 years with either recessive or dominant forms of inheritance (28, 29). The disease clinically appears much like SMA III, although it may be more progressive. The other form
is SBMA, or Kennedy’s disease, a sex-linked, recessive MND characterized by progressive spinal and bulbar muscular atrophy, gynecomastia, and reduced fertility (4, 30).
is SBMA, or Kennedy’s disease, a sex-linked, recessive MND characterized by progressive spinal and bulbar muscular atrophy, gynecomastia, and reduced fertility (4, 30).
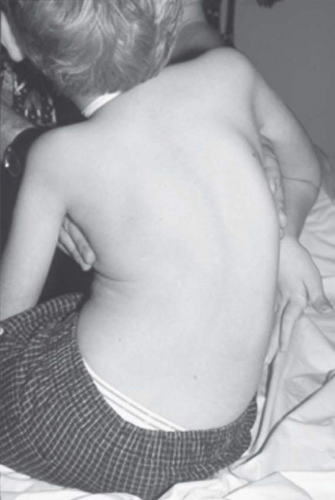 FIGURE 28-2. Scoliosis in a young boy with SMA type II. |
Adult-onset SMA, SBMA, and SMA III patients can have normal life spans, and many of the rehabilitative modalities discussed in this chapter are applicable to this population. Further, with the rapid advancement of rehabilitation technology, many SMA II patients are now living well into adulthood and successful pregnancies have been reported in this disease (31).
Genetics of SMA
A detailed analysis of the 5q13 region revealed that this chromosomal region in humans contained a large inverted duplication with at least two genes present in telomeric and centromeric copies. Further studies have identified the SMA causative gene as the survival motor neuron 1 gene (SMN1, telomeric copy), along with a disease modifying gene (SMN2, centromeric copy) (32, 33, 34, 35). Briefly, the two SMN genes are nearly identical except for a difference of only five nucleotides in their 3′regions, without any alteration of the amino acid sequence of the protein. However, the critical difference between SMN1 and SMN2 genes is a C-T transition located within the exon splicing region of the SMN2 that affects the splicing of exon 7. This change results in frequent exon 7 skipping during the splicing of SMN2 transcripts (32, 33). It is thought that the resulting truncated SMN protein without its exon 7 contribution is a less stable form of SMN protein and is therefore rapidly degraded. In about 95% of SMA patients, both copies of SMN1 exon 7 are absent due to mutations. In the remaining SMA affected patients, other small or subtle mutations have been identified (33, 34).
Genetic studies have now established that SMA is caused by mutations in the telomeric SMN1 gene, with all patients having at least one copy of the centromeric SMN2 gene. At least one copy of the SMN2 must be present in the setting of homozygous SMN1 mutations; otherwise, embryonic lethality occurs. The copy number of SMN2 varies in the population, and this variation appears to have some important modifying effects on SMA disease severity (34). It appears that higher number of SMN2 copies in the setting of SMN1 mutations results in less severe clinical SMA phenotype. However, substantial variations in SMA phenotype and disease severity can exist with a given SMN2 copy number, so it is not recommended to predict disease severity based on SMN2 copy numbers. Although we now know that SMN protein is expressed widely in many tissues throughout the body, its function is still not completely understood at this time (35).
Genetics of SBMA (Kennedy’s Disease)
SBMA is a hereditary adult-onset disease that causes preferential degeneration of LMNs leading to weakness and atrophy of bulbar, facial, and limb muscles. It is clinically similar to ALS. The main clinical distinction between the two is that ALS involves degeneration of both UMNs and LMNs, whereas the affected cell types in SBMA are LMNs. Another interesting difference to note is that Onuf’s nucleus, an androgen-sensitive spinal cord motor neuron nucleus, is spared in ALS, although it degenerates in SBMA (30, 35). Degeneration of sensory neurons of the dorsal root ganglia is also a typical sign associated with SBMA, often preceding the onset of motor dysfunction. In addition to the neurological phenotype, SBMA patients display some of the characteristic signs of androgen insensitivity syndromes including testicular atrophy, decreased fertility, gynecomastia, and elevated androgen levels (36). SBMA is caused by a novel mutation, the expansion of a trinucleotide CAG repeat, in the first exon of the androgen receptor gene (32). Unaffected individuals have a CAG repeat size that ranges between 5 and 35 glutamines, while symptomatic individuals always have a repeat size of at least 37 glutamines (32).
SBMA has some clinical variability; however, phenotypic expression does not correlate with the length of CAG repeats. This is in contrast to myotonic muscular dystrophy and fragile X syndrome, where increased numbers of tandem triplet repeats correlate directly with disease severity (30). Commercially available blood tests (DNA analysis) are now available for SMA and SBMA. SBMA can occur without any family history or gynecomastia and all males with atypical ALS should be tested for SBMA. Prevalence rates for SMA types II and III are estimated to range from as high as 40 per million among children to around 12 per million in the general population, with adult-onset SMA and SBMA being far less common (27).
DIAGNOSTIC EVALUATION OF MND
The diagnosis of ALS and other forms of adult MND is primarily a process of exclusion. If, based on the history and physical examination, clinical signs and symptoms of MND are detected, one must generate a differential diagnosis and then work to exclude processes mimicking MND. Only in FALS with known SOD1 mutations, Kennedy’s disease, and the few adult-onset SMA cases in which SMN mutations are detected is a definitive diagnostic test available. For most patients with ALS or its variants (PMA and PLS), electrodiagnostic testing (EDX), laboratory testing, neuroimaging studies, and, occasionally, a muscle biopsy are used to exclude other diagnoses. The El Escorial criteria (Fig. 28-3) are used to assess the certainty of a diagnosis of ALS once other disease processes have been excluded.
Clinical Presentation
Patients with ALS most often seek evaluation complaining of focal weakness (60%), rarely of generalized weakness or cramps, and very rarely of generalized fasciculations or respiratory failure. Symptom onset may be anywhere within the motor system, including areas of the brain and brainstem as shown in Figure 28-4. Although fasciculations are a prominent feature in most patients with ALS, patients who complain of fasciculations only and have an otherwise normal neurologic
examination usually have benign fasciculation syndrome and are unlikely to develop ALS. Symptom onset may be anywhere within the motor system.
examination usually have benign fasciculation syndrome and are unlikely to develop ALS. Symptom onset may be anywhere within the motor system.
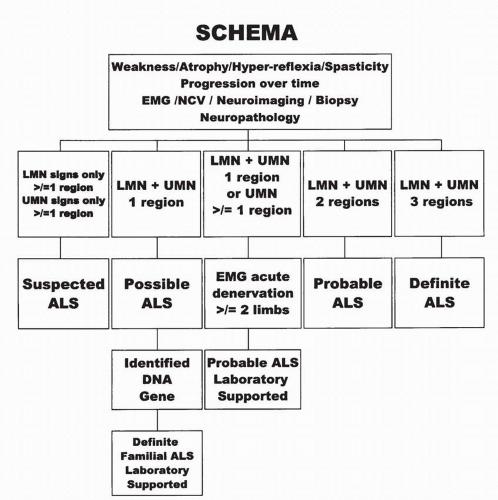 FIGURE 28-3. World Federation of Neurology El Escorial revisited criteria for ALS diagnosis. |
The evaluation of a patient suspected of having MND begins with a detailed history, general physical examination, and neurologic examination. On neurologic examination, one is looking for evidence of UMN and LMN dysfunction. The mental status, nonmotor cranial nerve function, sensory examination, and cerebellar examinations should be normal. The symptoms may be quite insidious and develop slowly or in unusual motor patterns. Loss of function may not be initially obviously disabling but performance may be affected. This is demonstrated by the noted decline in Lou Gehrig’s batting average over the last 10 years of his professional baseball career (Fig. 28-5).
Patients with UMN pathology often complain of loss of dexterity or a feeling of stiffness in the limbs. They may note weakness which is caused by spasticity resulting from disinhibition of brainstem control of the vestibulospinal and reticulospinal tracts. Findings on examination include spasticity and hyperreflexia, indicated by abnormal spread of reflexes and clonus or by the presence of brisk reflexes despite muscle atrophy due to LMN loss. The gold standard used to diagnose UMN pathology is the presence of pathologic reflexes, such as the Babinski’s sign, Hoffman’s sign, and jaw jerk. If the toe extensors are paralyzed, visualization of contraction of the tensor fascia lata when an attempt is made to elicit a Babinski response has the same significance as great toe extension. Recently, it has been suggested that the corneomandibular reflex may be a more sensitive and specific indicator than the jaw jerk of UMN pathology in the bulbar region (37).
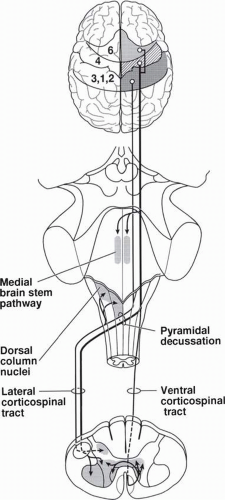 FIGURE 28-4. Areas of the brain and brainstem involved in ALS. |
Patients with LMN pathology usually present complaining of muscle weakness. In addition, they may note muscle atrophy, fasciculations, and muscle cramping. Cramping may occur anywhere in the body, including the thighs, arms, and abdomen. Cramping of abdominal or other trunk muscles raises a red flag urging the clinician to consider a diagnosis of ALS. Findings on examination include weakness, atrophy, hypotonia, hyporeflexia, and fasciculations. Head drop is a manifestation of muscle weakness often seen in ALS although it can be seen in other neuromuscular disorders; ALS and myasthenia gravis are the two most common causes of head drop. Atrophy often appears first in the hand intrinsic muscles. Although fasciculations are not a necessary criteria for the diagnosis of ALS, one should question the diagnosis when none are observed. A recent study identified the following hierarchy of initial symptoms: leg weakness in nearly half of ALS patients; followed by arms, bulbar muscles, and then generalized weakness (38). Presentation with only respiratory muscle weakness is extremely rare.
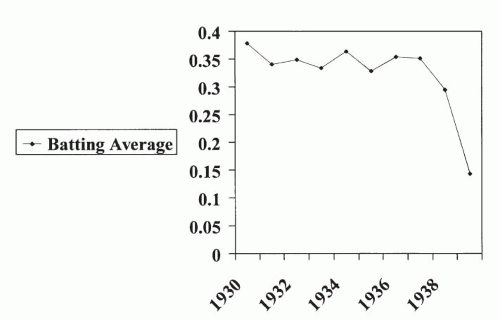 FIGURE 28-5. Batting average of Lou Gehrig during the last 10 years of his baseball career. |
Signs and symptoms suggesting bulbar muscle weakness include dysarthria, dysphagia, drooling, and aspiration. These signs and symptoms may be caused by UMN and/or LMN dysfunction involving the bulbar muscles. Signs of spastic dysarthria, indicating UMN pathology, include a strained and strangled quality of speech, reduced rate, low pitch, imprecise consonant pronunciation, vowel distortion, and breaks in pitch. LMN dysfunction creates a flaccid dysarthria in which speech has a nasal and/or wet quality; pitch and intensity are monotone, phrases abnormally short, and inspiration audible. Complaints of difficulty chewing and swallowing, nasal regurgitation, or coughing when drinking liquids, may all indicate dysphagia. On physical examination, the following tests may be used to assess facial and bulbar muscle function: ability to bury the eyelashes, pocket air in the cheeks, whistle; jaw opening and lip closure strength, phonation of a variety of syllables such as “puh,” “kuh,” “tuh,” and “ah.” The tongue should be examined for fasciculations and atrophy and tongue strength and range of motion assessed. The gag reflex and jaw jerk should be assessed to look for UMN dysfunction. Pseudobulbar affect is a symptom of pseudobulbar palsy which refers to an UMN syndrome caused by motor neuron loss in the corticobulbar tracts. Patients experience inappropriate laughter or crying which is not concordant with their mood and can be embarrassing. Disinhibition of limbic motor control produces pseudobulbar affect, also sometimes called “emotional incontinence.”
In patients who present with respiratory failure, the earliest signs are often nocturnal and include poor sleep with frequent awakening, early morning headaches, excessive daytime fatigue and sleepiness, nightmares, and orthopnea. Frequent sighing, a weak cough, and difficulty clearing bronchial or pulmonary secretions are other signs or respiratory muscle weakness. Later signs of respiratory dysfunction are dyspnea with exertion, truncated speech, respiratory paradox, dyspnea when eating, rapid shallow breathing, visible accessory muscle contraction, and flaring of the nasal alae. With advanced, untreated respiratory failure, patients may have an elevated hematocrit, low serum chloride, respiratory acidosis with a compensatory metabolic alkalosis, hypertension, and cor pulmonale.
Other signs and symptoms frequently associated with ALS are cachexia, fatigue, and musculoskeletal complaints. The term “ALS cachexia” refers to a phenomenon experienced by some patients in which weight loss occurs in excess of that caused by muscle atrophy and reduced caloric intake. Both subcutaneous fat and peritoneal fat are lost, presumably because of acceleration of the basal metabolic rate (39). In patients with ALS cachexia, greater than 20% of body weight is typically lost over a 6-month period. Many patients with ALS feel an overwhelming sense of muscle fatigue which is probably due to a combination of blocking of neuromuscular transmission in reinnervated nerve terminal sprouts and impairment of excitation contraction coupling (39). Some patients seek initial medical attention because of fractures or sprains that do not heal. In reality, these patients probably sustained their initial injury because of a fall or other injury (e.g., sprained ankle) that occurred because of underlying muscle weakness; they were then unable to recover to their premorbid level of function because of that weakness. Other common musculoskeletal complaints include neck and back pain, shoulder pain due to a frozen shoulder, elbow flexion and ankle plantar flexion contractures, and claw hand. Patients may experience osteoporotic fractures and/or stress fractures because of immobilizationinduced bone density loss.
Rare signs and symptoms which usually occur only in advanced ALS include sensory impairment, autonomic dysfunction, bowel and bladder dysfunction, extraocular muscle paralysis, pressure ulcer formation, and severe dementia. Although ALS is discussed as a pure motor disorder, some patients complain of paresthesias. These may be due to compression or entrapment neuropathies, but subclinical abnormalities in somatosensory evoked potentials and quantitative sensory testing have been reported (40).
Differential Diagnosis
After obtaining a history and examining the patient, the clinician is able to generate a differential diagnosis which guides further diagnostic testing. The differential diagnosis differs depending on whether the presentation is primarily LMN, UMN, bulbar or mixed LMN and UMN.
El Escorial Criteria
The El Escorial criteria (see Fig. 28-3) for diagnosing ALS were developed by a task force of the World Federation of Neurology in 1990 to ensure inclusion of more homogeneous patient populations in ALS clinical trials (41). These criteria have been used to enroll patients in most of the recent clinical trials. The criteria were revised in 1998 to improve the speed and certainty of diagnosis (42). The criteria classify the certainty level of the diagnosis of ALS as falling into one of five categories: definite, probable, probable with laboratory support, possible, and suspected. In brief, the motor system is divided into four regions: bulbar, cervical, thoracic, and lumbosacral. Clinical evidence of UMN and LMN pathology is sought in each region. The certainty level of diagnosis depends on how many regions reveal UMN and/or LMN pathology. Figure 28-3 summarizes the
schema for placing patients in the five diagnostic categories. Clinical weakness, atrophy, and fasciculations are considered evidence of LMN pathology. Pathologic spread of reflexes, clonus, and pseudobulbar features are considered evidence of UMN pathology. Electrophysiologic findings can be used to both confirm LMN dysfunction in clinically affected regions and to detect LMN dysfunction in clinically uninvolved regions. Neuroimaging and clinical laboratory studies are used to exclude other conditions that may mimic ALS.
schema for placing patients in the five diagnostic categories. Clinical weakness, atrophy, and fasciculations are considered evidence of LMN pathology. Pathologic spread of reflexes, clonus, and pseudobulbar features are considered evidence of UMN pathology. Electrophysiologic findings can be used to both confirm LMN dysfunction in clinically affected regions and to detect LMN dysfunction in clinically uninvolved regions. Neuroimaging and clinical laboratory studies are used to exclude other conditions that may mimic ALS.
Electrodiagnostic Testing
The various forms of MND, including SMA, Kennedy’s disease, PMA, SALS, and FALS share several electrodiagnostic features but also differ in some aspects due to varying rates of disease progression (43). General EDX characteristics of MND include normal sensory nerve conduction studies (NCS), normal or low motor amplitudes depending on disease stage, and normal distal motor latencies and conduction velocities. However, with profound loss of motor amplitude, conduction velocities may drop as low as 25% below the lower limit of normal because of loss of the fastest conducting fibers. The needle electrode examination (NEE) reveals a decreased recruitment pattern, either normal size or large motor unit action potentials (MUAPs) with or without evidence of remodeling depending on the specific disease process, and abnormal spontaneous activity including positive sharp waves (PSWs), fibrillation potentials, fasciculations, and complex repetitive discharges (CRDs). The prominence of the various forms of spontaneous activity varies with the different forms of MND.
Spinal Muscular Atrophies
The EDX features of the autosomal recessive SMAs I-IV are determined by the rate of anterior horn cell degeneration and the stage in the course of the disease. Sensory NCS are normal in all forms of SMA. Compound motor action potentials (CMAPs) are decreased in proportion to the degree of muscle atrophy. Motor velocities are most likely to be abnormally slow in SMA I because of the extensive loss of motor axons.
The most profound loss of MUAPs is seen in SMA I. With maximal effort, only a few MUAPs may fire at a rapid rate. Small MUAPs are common because reinnervation cannot compensate for the rapid loss of anterior horn cells. Myopathic appearing low amplitude, polyphasic, short duration units may also be seen because of muscle fiber degeneration. In the other types of SMA, one sees large amplitude MUAPs (up to 10 or 15 mV) because the number of fibers per motor unit increases as motor unit remodeling occurs. These large units also tend to be polyphasic with increased duration. Satellite potentials appear as remodeling occurs. Myopathic appearing MUAPs are also seen in some older patients with SMA III, and their etiology is not well understood.
On NEE in SMA I, fibrillation potentials and PSWs are diffuse and seen in many muscles, including the paraspinals. In the more chronic forms of SMA, fibrillation potentials and PSWs are even more common and increase in frequency as age increases. CRDs are often seen in SMA II and III, and fasciculations are more common than in type I (44, 45, 46).
Kennedy’s Disease
Motor NCS abnormalities are similar to those seen in other forms of MND. Although patients generally do not have sensory complaints, absence or reduction of sensory nerve action potential (SNAPs) is a common finding (47, 48). NEE shows large amplitude and duration MUAPs consistent with the rather indolent disease course. Fibrillation potentials and PSWs may be very prominent and present in all muscles examined. Fasciculation potentials are also abundant in limb, facial, and tongue muscles.
Adult Nonhereditary MND
For many years, Lambert’s criteria were the standard for the electromyographic (EMG) diagnosis of ALS (49, 50). The following four criteria were required to make a definite diagnosis of ALS: (a) PSWs and/or fibrillation potentials in three of five limbs, counting the head as a limb. For a limb to be considered affected, at least two muscles innervated by different peripheral nerves and roots should show active denervation. (b) Normal sensory NCS. (c) Normal motor conduction studies; however, if the CMAP amplitude is very low, conduction velocity may drop as low as 70% of the lower limit of normal. (d) Reduced recruitment of MUAPs on needle exam. More recently, Cornblath et al. studied 61 patients with ALS and found that even with low CMAPs, motor distal latencies, and F wave latencies did not exceed 125% of the upper limit of normal, and motor conduction velocities did not fall below 80% of the lower limit of normal (51). The EDX findings in PMA are identical to those in ALS; the distinction between the two diagnoses is made by the presence or absence of UMN signs on physical examination. By definition, the EDX examination is normal in PLS. In PBP, active denervation is found only in muscles of the head and neck.
The EDX portion of the El Escorial criteria differs somewhat from Lambert’s criteria and is generally more liberal. The revised El Escorial criteria allow EDX findings to be used to upgrade the certainty of a diagnosis from clinically possible ALS to probable ALS; this upgrading of the diagnosis is important because it often allows additional patients to participate in clinical trials which generally require a diagnosis of probable or definite ALS. The El Escorial EDX criteria state that active denervation must be present in two of the four spinal regions (bulbar, cervical, thoracic, and lumbar) to support a diagnosis of ALS. For the cervical or lumbosacral region to be counted, at least two muscles innervated by different nerve roots and peripheral nerves must have EMG changes. In the bulbar and thoracic regions, changes in one muscle are sufficient. Thus, a patient with active denervation in the left arm and thoracic paraspinals would meet the El Escorial criteria for an EDX diagnosis of ALS but not the Lambert criteria because only one limb is involved. On the other hand, a patient with denervation in the tongue and both arms would fulfill Lambert’s criteria and the El Escorial criteria for ALS because three limbs and two regions are involved (bulbar and cervical). Early in the
progression of ALS, many patients with a suspected clinical diagnosis do not meet EDX criteria for a definite diagnosis. A repeat study several months later will often fulfill the EDX criteria for diagnosis.
progression of ALS, many patients with a suspected clinical diagnosis do not meet EDX criteria for a definite diagnosis. A repeat study several months later will often fulfill the EDX criteria for diagnosis.
NCS in ALS are characterized primarily by decreased CMAP amplitudes. The mild slowing of motor conduction velocity and the prolongation of F wave latencies are attributed to loss of the fastest conducting axons. An interesting phenomenon observed in many patients is that of the “split hand”; CMAP amplitudes are decreased to a greater extent on the radial side of the hand than on the ulnar side. CMAPs obtained from the abductor pollicis brevis and first dorsal interosseous are much lower than those obtained from the abductor digiti minimi. More than two stimulation sites should be used in the evaluation of motor nerves to exclude the presence of conduction block since multifocal motor neuropathy with conduction block is occasionally misdiagnosed as ALS. The ulnar nerve easily can be stimulated at the wrist, below the elbow, above the elbow, in the axilla, and in the supraclavicular fossa. In limbs with UMN signs, H-reflexes may be elicited from muscles in which they cannot normally be obtained. A few patients do have SNAP abnormalities, and the sympathetic skin response is absent in 40%, suggesting subclinical autonomic nervous system involvement (52). Repetitive stimulation studies may show a decrement in CMAP with stimulation at 3 Hz which is similar to that seen in myasthenia gravis. A decrement is especially likely to be detected in patients with rapidly progressing disease and in muscles with an abundance of fasciculations (43).
The NEE is the most important part of the EDX examination in cases of suspected ALS. Fasciculation potentials are seen in most patients with ALS, but they are not necessary to meet diagnostic criteria, and the presence of only fasciculations is inadequate as evidence of LMN involvement of a particular limb or region. The significance of fasciculations depends on the company they keep; they are pathologic only when accompanied by fibrillation potentials, PSWs, or recruitment pattern or MUAP size changes. In patients with advanced ALS, fibrillation potentials and PSWs are prominent in most muscles, but they may be sparse early in the course of the disease. Occasionally, CRDs and doublets or triplets are seen in patients with ALS, but these are not typical EDX findings in ALS. The thoracic paraspinals should be examined with a needle; they are not involved in tandem cervical and lumbar stenosis and can help exclude this as a diagnostic possibility. In addition, when the El Escorial criteria are employed, the finding of denervation in the thoracic and in either the cervical or the lumbar region is sufficient for a definite diagnosis, making examination of the tongue or facial muscles, which many patients find unpleasant, unnecessary. Although fasciculations and denervation of the tongue are considered almost pathognomonic for ALS, they are seldom found in patients who do not have clinical evidence of bulbar muscle involvement. The recruitment pattern is decreased in involved muscles. If the disease is progressing relatively slowly, MUAP amplitudes and durations become increased; but if the course is very rapid, denervation outpaces reinnervation and enlarged MUAPs do not have time to develop. The density and distribution of fasciculations and fibrillations do not correlate with disease course or prognosis, and serial EDX examinations are not useful for monitoring disease progression once a definite diagnosis has been made.
Neurophysiologists have begun to explore the use of transcranial magnetic stimulation as a method of identifying subclinical UMN dysfunction. Results are contradictory with respect to the sensitivity and specificity of various findings as evidence of UMN dysfunction, and these techniques must be considered experimental at present. Abnormalities suggesting UMN pathology include a motor evoked potential (MEP) much lower in amplitude than the CMAP recorded from the same muscle, prolonged central motor conduction time, decreased MEP thresholds and silent periods early in disease, increased MEP thresholds in advanced disease, and decreased cortical representation of individual muscles (53, 54, 55, 56).
Neuroimaging
Imaging studies are used to exclude possibilities other than MND from the differential diagnosis. Magnetic resonance imaging (MRI) is the primary imaging modality used in the evaluation of patients with suspected ALS. Almost all patients should have an MRI of the cervical spine to rule out cord compression, a syrinx or other spinal cord pathology. The location of symptoms will dictate whether or not other regions of the spinal cord should be imaged. In patients presenting with the PMA phenotype, an MRI of the involved region of the spinal cord with gadolinium should be considered to look for a metastatic polyradiculopathy. In those presenting with bulbar symptoms, a brain MRI should be performed to rule out stroke, tumor, syringobulbia, etc.
Although MRI is generally not performed to confirm a diagnosis of ALS, a few associated abnormalities have been reported. Rarely, spinal cord and motor cortex atrophy is apparent. Corticospinal tract hyperintensity with T2 imaging has been observed in a few younger patients with a predominance of UMN signs (53).
Laboratory Evaluation and Other Diagnostic Tests
In most neuromuscular clinics, a routine panel of laboratory tests is performed for all patients suspected of having ALS. The rationale behind performing this battery of tests is to assess the general health of the patient and exclude treatable conditions. The differential diagnosis, developed following the history and physical exam, may suggest that more specialized testing be performed. Additional tests may be warranted when the presentation is with the PMA, PLS, or PBP phenotype. When there is a family history of MND, genetic testing for FALS is performed.
ALS CLINICAL TRIALS
Clinical trials play a crucial role in the development of new therapeutic agents. It is important to begin thinking about
what an appropriate trial would look like far in advance of actual study initiation. Decisions regarding dose, outcomes to be measured, and duration of treatment all may have a critical impact on whether a new agent is found to be efficacious. Many of these decisions depend on adequate preclinical data. In the absence of good preclinical data, trials may fail to show efficacy or even demonstrate harm to patients that could have been avoided. In recent years, much has been learned about pathogenic mechanisms of ALS, leading to a proliferation of new targets for disease modification. Mitochondrial dysfunction, glutamate toxicity, protein misfolding, and microglial activation are just a few mechanisms that have been proposed. For each proposed mechanism, pharmacological manipulation is possible. Targeted drug discovery programs can lead to new compounds, and reevaluation of existing drugs may lead to recognition of properties not previously investigated. Recently, a collaborative effort jointly funded by the National Institute of Neurological Disorders and Stroke (NINDS) and the Amyotrophic Lateral Sclerosis Association (ALSA) tested more than 1,000 available compounds in 29 different assays to determine activity against a variety of different aspects of neurodegeneration (57). Although the full result of this effort has not yet been published, individual laboratories have further investigated drugs identified by this screening program, with the first of these (ceftriaxone) having entered clinical trials in 2006. At this writing, there are at least nine different compounds either in human trials or about to be tested in humans. All of these compounds target different aspects of the neurodegenerative process.
what an appropriate trial would look like far in advance of actual study initiation. Decisions regarding dose, outcomes to be measured, and duration of treatment all may have a critical impact on whether a new agent is found to be efficacious. Many of these decisions depend on adequate preclinical data. In the absence of good preclinical data, trials may fail to show efficacy or even demonstrate harm to patients that could have been avoided. In recent years, much has been learned about pathogenic mechanisms of ALS, leading to a proliferation of new targets for disease modification. Mitochondrial dysfunction, glutamate toxicity, protein misfolding, and microglial activation are just a few mechanisms that have been proposed. For each proposed mechanism, pharmacological manipulation is possible. Targeted drug discovery programs can lead to new compounds, and reevaluation of existing drugs may lead to recognition of properties not previously investigated. Recently, a collaborative effort jointly funded by the National Institute of Neurological Disorders and Stroke (NINDS) and the Amyotrophic Lateral Sclerosis Association (ALSA) tested more than 1,000 available compounds in 29 different assays to determine activity against a variety of different aspects of neurodegeneration (57). Although the full result of this effort has not yet been published, individual laboratories have further investigated drugs identified by this screening program, with the first of these (ceftriaxone) having entered clinical trials in 2006. At this writing, there are at least nine different compounds either in human trials or about to be tested in humans. All of these compounds target different aspects of the neurodegenerative process.
Assuming that the maximum tolerated dose (MTD) has been established in experimental models, as well as a range of doses that show activity against the target disease mechanism, dose ranges in the initial studies on humans can be appropriately chosen. In ALS, this step has been problematic. For a number of compounds that have been previously studied in efficacy trials, the MTD has not been established. Thus, negative results have been reported for creatine and celecoxib, yet the lack of a known MTD leads to the question of whether higher doses of either creatine or celecoxib could have demonstrated efficacy (58, 59). In other studies, attempts were made to study compounds at doses close to MTD, but lower doses were not studied as well. This may have contributed to the fact that patients treated with topiramate at 800 mg/day progressed faster than placebo patients, and may also account for similar results in the recently reported minocycline trial (60, 61).
ALS presents some unusual challenges in terms of clinical trials. In diseases associated with markers of activity (i.e., CD4 counts or viral load in HIV), the effect of differing doses on these markers can be used to determine the dose choices for a phase III trial. In ALS, no such markers have been identified, so that attempts to gauge efficacy must be based on the outcomes typically employed in larger phase III trials. For this reason, the line between phase II and phase III trials is often blurred in ALS. Thus, decisions about dose are often made after phase II trials, so it is essential that multiple doses be evaluated. This has often not been done in phase II ALS trials, and when dose ranging is done, it is often inadequate. For example, topiramate was tested in a phase II study at a dose of 800 mg/day (62). Although there was no statistically significant effect on mortality, treated patients lost an average of 10 lb more weight than placebo-treated patients and performed more poorly on functional and respiratory measurements (62). From assessment of adverse events, it was clear that at this dose, topiramate was quite difficult to tolerate, and the results reported could easily have been a function of patient’s weight loss and other events.
Outcome Measures
Given that there are no tissue-based biomarkers currently existing to determine drug activity in ALS, clinical assessment of efficacy is based on measurement of a variety of aspects of disease. The gold standard outcome for ALS trials currently remains survival. Survival is obviously clinically meaningful and straightforward to measure. There are several reasons, however, why other measures are being sought and why many current trials use outcomes other than survival. First, survival can be manipulated by many interventions that do not clearly alter the progression of underlying disease. Good nutrition and early use of percutaneous endoscopic gastrostomy (PEG) clearly prolong life (63). Respiratory support with noninvasive positive pressure ventilation (NIPPV) has been less well studied, but likely also prolongs life (64, 65). Beyond these clearly defined interventions, there is emerging evidence that patients cared for at multidisciplinary ALS clinics have prolonged survival as compared to community-based controls (66, 67). As these interventions may not be applied uniformly across all sites in a clinical trial, conclusions based on survival may be confounded by these variables. Many trials stratify along certain treatment variables, but stratification can reduce the power of a trial to find a significant drug benefit.
PHARMACOLOGIC MANAGEMENT OF MND
Riluzole
Despite clinical use for more than 14 years, riluzole, a 2-amino-6-(trifluoromethoxy) benzothiozole, remains the only Food and Drug Administration (FDA)-approved medication proven to slow the progression of ALS. Pharmacological mechanisms of riluzole include interference with N-methyl D-aspartate (NMDA) receptor-mediated responses, stabilization of the inactivated state of voltage-dependent sodium channels, inhibition of glutamate release from synaptic terminals, and activation of extracellular glutamate uptake. Riluzole has demonstrated neuroprotective effect in motor neuron cultures and SOD1G93A transgenic mice (68, 69, 70, 71, 72, 73, 74, 75, 76




Related posts:





Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
