Chapter 12 Abnormalities in Immune Complex Clearance and Fcγ Receptor Function
The Role of the Mononuclear Phagocyte System in the Clearance of Immune Complexes
Early studies of the blood clearance of bacteria in mice, rabbits, and guinea pigs demonstrated that the mononuclear phagocyte system performed this function for opsonized particles. Infused bacteria were internalized by hepatic and splenic phagocytes.1 The rate of clearance of bacteria from the blood and the site of their clearance depended on the level of antibodies to the bacteria in the serum of the animal. Rapidly cleared, well-opsonized bacteria were principally phagocytosed in the liver, whereas the more slowly cleared, less efficiently internalized (and presumably less opsonized) bacteria were removed by splenic phagocytes. These observations are remarkable for their similarity to the models of immune complex clearance in animals and humans that are described later.
Animal models have shown that the mononuclear phagocyte system serves an important role as a site for removal of soluble immune complexes.2,3 This system may be saturated with increasing amounts of infused immune complexes, resulting in glomerular deposition of complexes, as seen in SLE.4 Although impairment of immune complex clearance leads to increased deposition in tissues, the absence of activating FcγR on phagocytes prevents an inflammatory response to the localized immune complexes. Mice with targeted deletions of activating FcγR are protected from fatal antigen-antibody Arthus reactions and immune complex–mediated glomerulonephritis. In contrast, mice lacking inhibitory FcγRs have exaggerated responses to immune complexes.5–7
Animal models of endogenous immune complex deposition also support the relationship between depressed mononuclear phagocyte system clearance and the genesis of glomerulonephritis. In chronic serum sickness, there is decreased clearance of aggregated albumin8 and aggregated human immunoglobulin G (IgG).9 Decreased clearance of heat-aggregated IgG in murine nephritis10 and of polyvinyl pyrrolidine in New Zealand black/white (NZB/W) mice11 has been observed, although some studies of endogenous immune complex–mediated disease have not found dysfunction of the mononuclear phagocyte system. The principle to be derived from these animal models of immune complex disease, whether from infused immune complexes or endogenous disease, is that immune complex deposition is influenced by the efficiency of mononuclear phagocyte system clearance. Specifically, impairment of mononuclear phagocyte system clearance is associated with tissue deposition of immune complexes and the potential for local organ damage.
Mechanisms of Immune Complex Clearance
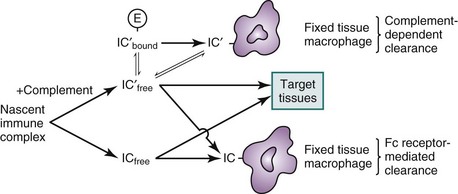
A number of factors govern the physical characteristics of immune complexes and, hence, their biologic properties (Box 12-1). These include the nature of the antibody in the complex, the nature of the antigen, and the antigen-antibody interaction. Antigen and antibodies in the circulation may rapidly form immune complexes, but the immunochemical properties of these circulating immune complexes determine their ultimate fate, either removal by the mononuclear phagocyte system or deposition in tissues. The potential of immune complexes to interact with FcγRs, to fix complement, and to react with complement receptors influences their rate of clearance. Immune complexes without complement are cleared primarily by FcγRs on fixed-tissue macrophages. Complexes that are opsonized with sufficient complement may bind to the receptor for C3b on circulating erythrocytes and subsequently may be removed by FcγRs and complement receptors. Thus, two classes of receptors, the FcγRs on phagocytes and the complement receptors on both erythrocytes and phagocytes, participate in the clearance of immune complexes (Figure 12-1).
Complement Mechanisms: Immune Adherence and the Erythrocyte CR1 System
Complement component 3 and the receptor for C3b on erythrocytes are important in processing and transporting large immune complexes12 (see Chapter 13). Incorporation of complement components, C3b in particular, modifies the solubility of large immune complexes13,14 and mediates the binding of immune complexes to human and other primate erythrocytes. Although both the liver and spleen are the major sites of immune complex uptake, erythrocytes in primates12,15 and platelets in rodents16,17 are important in clearing/processing immune complexes from the circulation. It has long been known that large complement-opsonized immune complexes bind to human erythrocytes.18 Termed immune adherence, this reaction has been shown to participate in the handling of nascent circulating immune complexes in primates.19
Human erythrocytes express complement receptor type 1 (CR1), which permits binding of complement-fixing immune complexes. CR1 on erythrocytes can be conceptualized as having three main functions, which are not mutually exclusive: buffering, transporting, and processing (see Figure 12-1). The role of immune complex buffer has been suggested for erythrocytes because erythrocyte-bound immune complexes are unavailable for tissue deposition but nonbound complexes can deposit in the tissues. Bound immune complexes are transported to the liver or spleen, where fixed-tissue phagocyte FcγRs and complement receptors strip the immune complexes from the erythrocytes, which then return to the circulation to continue this process, thus performing the transporting function. Finally, CR1 promotes degradation of captured C3b on immune complexes, thereby modifying their subsequent handling.
The human CR1 (the complement receptor for C3b/C4b and, to a lesser degree, iC3b) is a single-chain, intrinsic membrane glycoprotein expressed on several different cells, including erythrocytes, granulocytes, monocytes, and macrophages (see Chapter 13). There are four codominantly expressed alleles of CR1, with molecular weights of 220,000, 250,000, 190,000, and 280,000 daltons (Da).20–23 Inherited and acquired differences in the numeric expression of CR1 on erythrocytes have been described and associated with SLE.24–28 Two alleles with codominant expression determine erythrocyte CR1 number in healthy individuals.27,29 Although the CR1 number expressed on erythrocytes is low compared with that on leukocytes, approximately 90% of total circulating CR1 is on erythrocytes, because there are far more erythrocytes than leukocytes in the circulation.30,31
The binding of immune complexes to CR1 occurs rapidly in vivo, and it represents multivalent binding between multiple C3b molecules on the complex and clusters of CR1 on erythrocytes.19,32–35 In vivo studies have demonstrated that immune complexes preferentially bind to circulating erythrocytes that express multiple CR1 clusters and that the capacity of each erythrocyte for binding correlates with the density of cell surface CR1. Because CR1 on erythrocytes tends to cluster more than that on resting neutrophils, most immune complexes that are bound to circulating cells are bound to erythrocytes.13,15,30,31,36–40 A reduction in the number of functional CR1s limits the capacity of erythrocytes to transport and buffer immune complexes, and in vivo studies have demonstrated that repeated administration of antigens in immunized humans and primates with immune complex formation results in a decrease in erythrocyte CR1 levels.36,41 Studies with primates have suggested that circulating immune complexes that are not bound to erythrocytes are more easily trapped in the microvasculature and can be recovered in the lungs and kidneys.40,42 Taken together, these findings have obvious implications for immune complex–mediated diseases.
The erythrocyte CR1 system may also have a second physiologic function: providing a processing mechanism for immune complexes.43 In addition to being a carrier for opsonized immune complexes, CR1 has a potent inhibitory function in the complement cascade, a function that may enhance clearance. It participates in the inactivation of C3b and may alter the size of complexes, thus affecting their subsequent handling. Specifically, CR1 is a cofactor for factor I in the cleavage of C3b to iC3b and then to C3dg.44,45 Therefore, the binding of immune complexes containing C3b to erythrocyte CR1 facilitates proteolytic cleavage of the C3b to iC3b and C3dg, which do not bind to CR1. This reaction is the basis for the degradation of complement on immune complexes with their subsequent release from the receptor,46 and its rate varies with the physicochemical properties of the individual complexes.47 If the immune complex can again activate complement and bind C3b, it can rebind to CR1.48 The fraction of immune complexes in whole blood that is erythrocyte bound depends on several dynamic processes: complement fixation and C3b capture, erythrocyte binding, and C3b degradation and immune complex release.
Fcγ Receptor Mechanisms
Immune complexes are removed from the circulation by the mononuclear phagocyte system of the liver and spleen through engagement of FcγRs and complement receptors. The interaction of immune complexes with the phagocyte involves a qualitatively different process from that with erythrocytes.37 The relative contribution of each receptor system depends on the immunochemical properties of the complex. The liver, which is much larger than the spleen, is the principal site for the uptake of immune complexes42,49,50; however, immune complexes that escape clearance by hepatic macrophages, which may be smaller and of lower valence, are taken up by the spleen.49 The role of FcγRs in clearance of both soluble and particulate immune complexes is shown by studies wherein blockade of FcγRs by an infusion of aggregated IgG into the portal venous system30 or of antibodies against FcγRs37 suppresses uptake of these immune complexes (Figure 12-2). Supporting the pivotal role of FcγRs in handling certain immune complexes, studies of complement depletion show no effect on the efficiency of uptake of immune complexes by the liver or spleen and actually show an acceleration in the rate of removal of complexes from the circulation, presumably resulting from their being trapped in the microvasculature.40
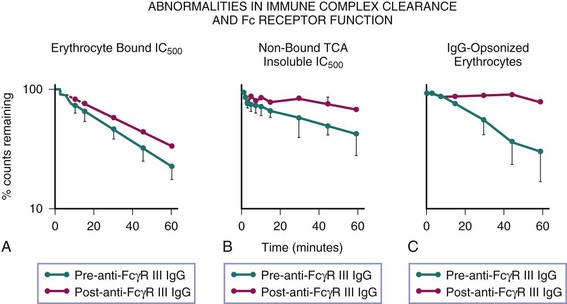
FcγRs appear to play a key role in the transfer and retention of immune complexes by mononuclear phagocytes. Studies of DNA/anti-DNA complexes that are bound to radiolabeled erythrocytes and injected into chimpanzees show that whereas immune complexes are removed by the mononuclear phagocyte system, the erythrocytes are not sequestered; rather, they are stripped of immune complexes and promptly recirculated.15 Although the mechanism of this stripping is not well defined, the involvement of complement proteases has been implicated.51 In this model of immune complex clearance, infusion of erythrocyte-bound DNA/anti-DNA complexes after treatment with anti-FcγR monoclonal antibody results in a significant amount of non–erythrocyte-bound circulating immune complexes, documenting the participation of FcγRs in the retention of immune complexes by phagocytes (see Figure 12-2B).37
In addition to stripping erythrocyte-bound complexes, FcγRs as well as CR3/CR4 are responsible for the clearance of those complexes that are unable to bind to erythrocyte CR1 because of inadequate C3b capture or degradation of C3b. This interpretation is supported by experiments in which primates treated with anti-FcγR monoclonal antibodies showed impaired clearance of infused immune complexes, which was most pronounced in the fraction of complexes that did not bind to erythrocytes.37 It has been shown that immune adherence is not a prerequisite for the efficient handling of immune complexes by the mononuclear phagocyte system,41 but immune complexes that do not fix complement or that fix complement poorly cannot be cleared if FcγR function is impaired (see Figure 12-1).
Abnormal Immune Complex Clearance in SLE
Analysis of the Clearance of IgG-Sensitized Autologous Erythrocytes
The technique introduced by Frank52 to measure mononuclear phagocyte system function employs autologous chromium Cr 51–labeled erythrocytes that are sensitized with IgG anti-(Rh)D antibodies and injected into study subjects, and clearance or removal of these cells from the circulation is determined by serial bleeding. External surface counting of sensitized radiolabeled erythrocytes shows initial rapid sequestration in the liver, followed by splenic accumulation of most of the injected cells. The semilogarithmic plot of mean data for the clearance of sensitized cells in normal control subjects is curvilinear, with a rapid initial loss of radiolabeled cells followed by a slower, sustained loss of radioactivity (Figure 12-3).53–55
 –
– , inactive/nonrenal disease (n = 5); •–• active/nonrenal disease (n = 7);
, inactive/nonrenal disease (n = 5); •–• active/nonrenal disease (n = 7); 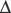 –
– , active/nonrenal disease (n = 12);
, active/nonrenal disease (n = 12);  –
– , active/renal (n = 8).
, active/renal (n = 8).Although originally conceptualized as a measure of FcγR capacity, kinetic analysis of in vivo clearance studies and in vitro studies with IgG anti-(Rh)D–coated erythrocytes suggests that complement also plays a role in clearance of this probe.56 A proposed model to describe the series of steps in handling of IgG anti-(Rh)D–sensitized erythrocytes is as follows: Circulating cells initially sequestered by a complement-dependent process are deactivated and released back into the circulation or are phagocytosed. Released cells are sequestered and phagocytosed by an FcγR-mediated process. Circulating cells may also be directly sequestered and phagocytosed by FcγRs.54–56
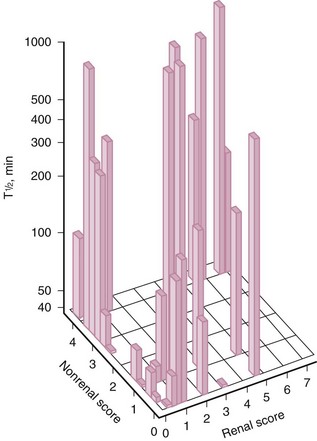
Abnormal mononuclear phagocyte system function in patients with SLE has been demonstrated in several studies performed with IgG anti-(Rh)D–sensitized erythrocytes.52,57–60 Clearance half-times for radiolabeled autologous IgG-sensitized erythrocytes were longer in these patients than in normal individuals and longer in patients with renal disease than in those without renal disease (Figures 12-3 and 12-4). When clinical activity in patients with SLE was assessed, there was a significant but independent association between impaired FcγR clearance and the level of both renal and nonrenal disease activity.59 Increased activity along either parameter was associated with more impaired clearance (see Figure 12-4). Longitudinal studies in patients with SLE showed that mononuclear phagocyte system function changed concordantly with changes in clinical status, indicating that clearance dysfunction is dynamic and closely related to disease activity.57,58
Analysis of Clearance of Infused Soluble Immune Complexes
As another measure of mononuclear phagocyte system function, the clearance of preformed, large, soluble, complement-fixing immune complexes has been studied in humans. Radiolabeled tetanus toxoid/antitetanus toxoid, hepatitis B surface antigen/antibody, or aggregated human IgG is infused, and then sequential blood samples are obtained and analyzed for whole blood and erythrocyte-bound radioactivity to monitor clearance.38,61,62 Clearance of these preformed immune complexes (free or erythrocyte bound) from the circulation of humans has been shown to involve the activation of complement with capture of C3b, binding to erythrocyte CR1 receptors, uptake by complement, and FcγR tissue mononuclear phagocytes, as described earlier. Factors that cause the erythrocyte transport system to fail, such as hypocomplementemia and CR1 deficiency, are associated with an initially more rapid disappearance of immune complexes, presumably caused by trapping in capillary beds outside the mononuclear phagocyte system. Given the different kinds of information obtained from each of these in vivo probes, examination of multiple models of immune complex clearance is necessary to define the mechanisms of immune complex deposition in SLE.
In vivo studies of infused soluble immune complexes complement the sensitized erythrocyte model of clearance and demonstrate multifactorial mononuclear phagocyte dysfunction. Abnormalities in the erythrocyte CR1 system, the early buffer for circulating immune complexes, are described in patients with SLE, and for these models it is important to recognize that such patients tend to have an acquired, decreased numeric expression of CR1 on erythrocytes that correlates with disease activity63,64 and may result from repeated immune complex/erythrocyte CR1 interactions.38,41 There also is evidence of an inherited deficiency of CR1 in some patients.26,28 With these model immune complexes, a rapid first phase of elimination was noted in patients with low complement, low CR1, and low immune adherence, which was ascribed to inappropriate tissue deposition of complexes.39 The second, slower elimination phase of infused aggregated IgG is also abnormal in SLE, presumably because of impaired splenic uptake as well as generalized mononuclear phagocyte dysfunction. Regardless of the mechanism, abnormalities in both splenic and hepatic clearance functions allow for a spillover of complexes beyond the mononuclear phagocyte system in SLE.
Blockade of FcγRs by elevations of IgG interferes with this key mechanism for the elimination of soluble circulating immune complexes.65,66 That serum concentrations of IgG are an important factor predicting the rate of aggregated IgG clearance in SLE67 emphasizes the importance of FcγR mechanisms in this model and supports the conclusions derived from the sensitized erythrocyte model of immune complex clearance. Specifically, FcγR-mediated clearance efficiency is crucial in SLE because of the defects in complement-dependent function.
Biology of Human Fcγ Receptors
Structure and Distribution
FcγRs are an essential receptor system that is engaged by immune complexes as they trigger internalization, release of inflammatory mediators, cytokines, and degranulation. In contrast to complement receptors, FcγRs recognize ligand in its native form. In humans, there are three distinct but closely related families of FcγRs—FcγRI (CD64), FcγRII (CD32), and FcγRIII (CD16)—that share many immunochemical and physicochemical properties and cellular distribution, and have highly homologous DNA sequences.68–72 Each of the eight FcγR genes leads to protein products with some unique features, including differences in binding capacity, distinct signal transduction elements, and cell-specific expression patterns (Figure 12-5). The structure-function relationships of each receptor family provide a framework for understanding how FcγRs may contribute to disease susceptibility, pathogenesis, and therapeutic intervention in SLE.
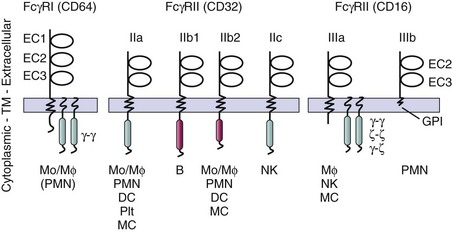
Both stimulatory and inhibitory FcγRs are often coexpressed on surfaces of hematopoietic cells, providing a mechanism to modulate cell activation initiated by stimulatory FcγRs. Studies have suggested that inhibitory FcγRs, which modulate thresholds for activation and can terminate activation signals, are a key element in the regulation of effector function.6,7 Inhibitory FcγRs play a central role in afferent and efferent immune responses as negative regulators of both antibody production and immune complex–triggered activation.
FcγRs belong to the immunoglobulin supergene family and are encoded by multiple genes on the long arm of chromosome 1q21-23.73–75 The presence of multiple distinct genes (arising from gene reduplication) and alternative splicing variants leads to a variety of receptor isoforms that are most strikingly different in transmembrane and intracellular regions, whereas they share similar but not precisely identical extracellular domains (see Figure 12-5).
FcγRs capable of triggering cellular activation possess intracellular activation motifs, termed immunoreceptor tyrosine-based activation motifs (ITAMs), similar to those of B-cell receptors and T-cell receptors.76,77 Inhibitory FcγRs have extracellular domains that are homologous to their activating counterparts, but their cytoplasmic domains contain an immunoreceptor tyrosine-based inhibitory motif (ITIM). The stimulatory FcγRI, a high-affinity receptor for IgG that binds monomeric IgG, and FcγRIIIa, an intermediate-affinity receptor that binds only multivalent IgG, are multichain receptors composed of a ligand-binding α-subunit, which confers ligand specificity and affinity, and associated signaling subunits with ITAMs in their respective cytoplasmic domains (Figure 12-5). FcγR α-chains are transmembrane molecules that share the structural motif of two or three extracellular immunoglobulin-like domains but vary in their affinity for IgG and in their preferences for binding different IgG subclasses (IgG1, IgG2, IgG3, and IgG4). Allelic variations in the ligand-binding regions of specific FcγRs influence the ability to bind certain IgG subclasses and alter the responses of phagocytes to IgG-opsonized antigens.78–80 The transmembrane domains of the α subunits contain a basic residue, which mediates the physical interaction with associated signal transducing chains required for efficient expression and signal transduction. Homodimeric γ-chains are transducing modules for FcγRI and FcγRIIIa (see Figure 12-5). Heterodimers of γ-ζ chains or homodimers of ζ chains can also transduce signals through FcγRIIIa in human natural killer (NK) cells. Another isoform, FcγRIIIb, has neither an ITAM nor a transmembrane domain, but is maintained in the plasma membrane outer leaflet by a glycosyl phosphatidylinositol (GPI) anchor (see Figure 12-5). In addition to multichain receptors, there are two other types of activating FcγRs and one inhibitory receptor with two different splice variants. FcγRIIa and FcγRIIc are single-chain receptors that include an extracellular ligand-binding domain and an ITAM in the cytoplasmic domain. Inhibitory FcγRs, FcγRIIb1 and FcγRIIb2, are single-chain receptors with extracellular domains highly homologous to their activating counterparts and cytoplasmic domains with ITIMs (see Figure 12-5).81
FcγRI (CD64) is distinguished by three extracellular immunoglobulin-like domains, a relatively high affinity for IgG,82 and the capacity for binding monomeric IgG (see Figure 12-5).83–85 FcγRIa, a heavily glycosylated 72-kDa protein, associates with homodimers of the Fc common γ-chain, which also can associate with the high-affinity receptor for IgE (see Figure 12-5).86 FcγRIa is present on monocytes, macrophages, and myeloid-derived dendritic cells.87 Monocyte expression of FcγRI is markedly enhanced by interferon-γ (IFN-γ),88,89 and neutrophils that do not constitutively express FcγRI can be induced to express this receptor by IFN-γ and granulocyte colony-stimulating factor (G-CSF).90,91
The FcγRII (CD32) family contains three genes encoding receptors that have low affinity for IgG and interact only with multimeric IgG in complexes. CD32 family gene products are the most widely expressed FcγRs and are found on most leukocytes and platelets.92–94 Density of expression varies with cell type but generally is higher than that for FcγRI.95 The structural heterogeneity of FcγRII family reflects three genes, FCGR2A, FCGR2B, and FCGR2C, which encode FcγRIIa, FcγRIIb, and FcγRIIc proteins, respectively, as well as several splice variants (see Figure 12-5).69–7296 With near identity in their extracellular and transmembrane domains, the gene products show divergence in cytoplasmic tails, which determines the effector functions that are mediated by each isoform (see Figure 12-5). Among the FcγRII family members, there are activating and inhibiting receptors that differ mainly in the signaling motif in the cytoplasmic domain. FcγRIIa and FcγRIIc contain ITAMs and they are preferentially expressed on cells of myeloid lineage, monocytes, neutrophils, certain dendritic cells, platelets, and NK cells. In addition to different isoforms, there are two allelic forms of FcγRIIa (R131 and H131), which are expressed codominantly and have differing IgG subclass–binding specificities and functional capacities (discussed later).70,78,79
Although ITAMs can assume an inhibitory function is some special circumstances, FCGR2B is the only FCGR gene encoding an ITIM, the canonical motif for an inhibitory receptor (see Figure 12-5).81 A single-chain low-affinity receptor with extracellular domains highly homologous to FcγRIIa and FcγRIIc, FcγRIIb has two alternative splice isoforms, FcγRIIb1 and FcγRIIb2, which differ only in their intracytoplasmic regions.96 FcγRIIb1 contains an insertion of 19 amino acids that alters intracellular targeting pathways. Neither isoform can trigger cell activation. Instead, both isoforms of FcγRIIb, when coaggregated with ITAM-bearing receptors, are negative regulators of activation. In addition, FcγRIIb2 participates in endocytosis of multivalent ligands by phagocytes and antigen-presenting cells, and the intracytoplasmic insertion in FcγRIIb1 inhibits internalization.97 FcγRIIb can modulate tyrosine phosphorylation–based cell activation by stimulatory FcγR, B-cell receptor (BCR), T-cell receptor (TCR), Fc receptors for IgE,98 Toll-like receptors (TLRs), and others. However, to inhibit cell activation, FcγRIIbs are typically coclustered with the other activating receptors.99 For example, FcγRIIb coaggregation with FcγRIIa by IgG-opsonized particles blocks phagocytosis, and FcγRIIb coligation to BCRs by antibody-antigen complexes inhibits B-cell proliferation and antibody production.100,101 Thus, FcγRIIb-mediated negative regulation of ITAM-dependent cell activation endows IgG-containing immune complexes with the capacity to regulate B cells and inflammatory cells. Because activating and inhibitory FcγRs are often coexpressed, the balance between stimulatory and inhibitory inputs determines cellular response. Allelic polymorphisms have been described in FcγRIIb; those in the transmembrane domain alter inhibitory function in B cells, and those in the promoter region alter receptor expression.102–104
The low-affinity FcγRIII (CD16) family contains two proteins, each with cell type–specific expression and each encoded by distinct, yet highly homologous genes (FCGR3A and FCGR3B).70,71,105 FcγRIIIa is the most abundant FcγR on tissue-specific macrophages and thus is a key receptor of the mononuclear phagocyte system. It is present at high density on Kupffer cells in the liver and on macrophages in the spleen, both important areas for immune complex clearance binding and internalization. In addition, it is expressed on dendritic cells, NK cells, γ/δ T cells, and mesangial cells.106 Different glycoforms of FcγRIIIa, with different affinities for IgG, are expressed on NK cells and macrophages.107 The FcγRIIIa α-chain is most typically associated with the Fc common γ-chain, a member of the family of signal transduction molecules that bear ITAMs within their cytoplasmic domains (see Figure 12-5).108 These signal transduction partners also are used by FcγRI, the high-affinity receptor for IgE and the T-cell receptor/CD3 complex. These accessory molecules form disulfide-linked dimeric complexes (homodimers or heterodimers) that noncovalently associate with the transmembrane region of FcγRIIIa to enable cell surface expression and signal transduction. FcγRIIIa also has the capacity to associate with the β-subunit of the high-affinity IgE receptor.109
FcγRIIIb is expressed at high levels on neutrophils and is the most abundant FcγR in the circulation. As a GPI-anchored receptor, it differs from FcγRIIIa, which is expressed on macrophages and NK cells as a conventional transmembrane protein (see Figure 12-5).110,111 FcγRIIIb on the surfaces of neutrophils interacts with β integrin CD11b/CD18, a finding of interest because of the strong genetic association of CD11b/ITGAM (integrin alpha M) locus and SLE. Further diversity in FcγRIII structure is provided by an allotypic variation in FcγRIIIb. The two most commonly recognized allelic forms of the GPI-anchored neutrophil isoform of FcγRIIIb, termed NA1 and NA2, differ by several amino acids and N-linked glycosylation sites.112,113 The alleles are inherited in a classic mendelian manner and are expressed in a codominant fashion. In addition to different isoforms, there are two allelic forms of FcγRIIIa (F176 and V176), which differ (1) in one amino acid at position 176 in the extracellular domain (phenylalanine and valine, respectively)80,114,115 and (2) in binding capacity for IgG1 and IgG3 (discussed later).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


