Metabolic Diseases, Nutritional Disorders, and Skeletal Dysplasias
ROBERT A. CHRISTMAN, JOHN TASSONE JR, AND CRISTINA MARCHIS-CRISAN
Many systemic disorders demonstrate associated radiographic findings in the lower extremity. Although these conditions typically are not clinically diagnosed by their foot or ankle manifestations, the practitioner should be familiar with their radiologic presentations so that they are not misdiagnosed as other conditions. The metabolic diseases, in particular osteoporosis, include several endocrine and nutritional disorders (hyperparathyroidism, acromegaly, scurvy, and rickets, for example). Paget disease may be localized in the lower extremity. Skeletal dysplasias present characteristic presentations that may be recognized as incidental findings; examples include osteopetrosis, osteopoikilosis, osteochondromatosis, and osteogenesis imperfecta. Also, soft tissue manifestations of systemic disorders are frequently encountered in the foot and leg.
On the other hand, recent literature shows not only that the bone is a target for hormones influencing calcium and phosphorus homeostasis and bone structure, but also that the bone itself produces at least two hormones (fibroblast growth factor 23 [FGF23] and osteocalcin), indicating the bone’s role as an endocrine organ. FGF23, produced by osteocytes in bone, act on the kidney to inhibit 1-alpha-hydroxylation of vitamin D and promote phosphaturia. Osteocalcin, an osteoblast product, acts on pancreatic beta cells to increase insulin production, glucose utilization, and visceral fat reduction.1
The pathology discussed in this chapter is organized according to its primary radiologic finding, which is useful when formulating differential diagnoses (Table 21-1).2
GENERALIZED OSTEOPENIA
Osteopenia
Decreased bone density, or increased radiolucency of bone, is a prominent radiographic feature of numerous metabolic disorders.3,4 Misconceptions abound regarding the use of terms relating to this finding5; therefore, the following definitions are strictly adhered to in this chapter. The term osteopenia, which means “poverty of bone,” is used to refer to the nonspecific radiographic finding of decreased bone density.3 The terms decalcification, undermineralization, and demineralization are not used, because they refer more specifically to the underlying physiology and pathology of bone as opposed to a descriptive radiographic appearance.6 Moreover, use of the term osteoporosis is reserved for the clinical entity or diagnosis.
Osteopenia occurs when bone resorption exceeds bone formation.7 When nearly one-third (30%–50%) of the bone mass is lost, radiographs can reveal osteopenia; this leads to defective bone quality and strength, causing a marked risk of osteoporosis.8
| Organization of Metabolic Disease, Endocrine and Hematologic Disorders, and Skeletal Dysplasias according to Primary Radiologic Findings in the Lower Extremity |
Regional osteopenia | Disuse and immobilization, complex regional pain syndrome |
Generalized osteopenia | Osteoporosis, osteomalacia, hypophosphatasia, hyperparathyroidism, renal osteodystrophy |
Epiphyseal region abnormality | Scurvy, rickets |
Altered bone architecture | Paget disease, fibrous dysplasia, sickle cell anemia, thallesemia |
Altered bone form | Acromegaly, gigantism, hereditary multiple exostoses, enchondromatosis, osteogenesis imperfecta, hypoparathyroidism, Albright hereditary osteodystrophy |
Generalized sclerosis | Osteopetrosis, osteopoikilosis, melorheostosis, osteopathia striata, pyknodysostosis, fluorosis, hypervitaminosis D |
Generalized periostitis | Hypertrophic osteoarthropathy, venous stasis, hypervitaminosis A, thyroid acropachy, tuberous sclerosis |
Chronic osteopenia can be a highly subjective radiographic feature; one must be careful with this “finding” because radiographic technique and/or processing can profoundly influence the visual appearance (i.e., the darkness or lucency) of bones. For example, a dark radiograph can result from increased kVp technique, mAs technique, developer time, or developer temperature. Instead, observe specific, more objective features in the forefoot.
Cortical and cancellous bone resorption is best identified using the dorsoplantar (DP) foot view and concentrating on the second, third, and fourth metatarsals. Patterns associated with cortical bone thinning in chronic osteopenia include endosteal resorption, intracortical tunneling (resorption along the surfaces of the Haversian canals), and/or subperiosteal resorption (Figure 21-1).3 A cancellous bone pattern associated with chronic osteopenia is prominent primary trabeculations, which may stand out in relief because secondary trabeculae are resorbed and subsequent bone is laid down on the remaining primary trabeculae (Figure 21-2). Abnormal cortical and cancellous bone findings can be seen individually or concomitantly (Figure 21-3). The medial oblique view should not be used for assessing osteopenia; striations that mimic intracortical tunneling normally are seen at the diametaphyseal region.
In contrast, the primary radiographic feature of acute osteopenia is spotty (mottled or moth-eaten) loss of bone density, particularly in periarticular regions (Figure 21-4). Acute osteopenia is associated with causes of regional osteoporosis, such as immobilization or disuse.
Osteoporosis
Osteoporosis is a metabolic disease that accompanies many other disease processes, including most of the endocrine and nutritional disorders discussed in this chapter. It is a condition in which the amount of bone present per unit of volume is reduced, but the composition is normal. Osteoporosis is characterized by progressive loss of bone mass, due to increased resorption as well as decreased bone production; a variety of disorders may have no commonalities except that they cause osteoporosis. Not only is there decreased bone mass, but also there is deterioration of the microarchitecture of bone tissue, such that bone becomes more fragile and is susceptible to fracture.9
Osteoporosis can also be subdivided based upon its cause: primary, affecting 80% of women and 60% of men, and secondary, affecting 20% of women and 40% of men. Primary osteoporosis, which mainly affects trabecular bone, can be further divided into two categories: (1) type I osteoporosis, due to estrogen withdrawal effect, occurring in postmenopausal women (age 51–75 years); and (2) type II osteoporosis (involutional or senile), frequently seen in both men and women of more than 70 years of age, and associated with fractures of the proximal tibia, femoral neck, and pelvis. Primary osteoporosis has a multifactorial etiology, with genetics, peak bone mass, nutrition, level of physical activity, age of menopause, and estrogen status playing a role. Secondary osteoporosis, which affects both trabecular and cortical bone, exists as a common feature of another disease, and as such, has an identifiable cause, such as hyperthyroidism, hyperparathyroidism, acromegaly, diabetes mellitus, gastrointestinal disease, malnutrition, and the drug side effects of corticosteroids.10
There are two forms of idiopathic osteoporosis based on age. Idiopathic osteoporosis, of unknown pathogenesis, has been described in middle-aged men; bone turnover is rapid, often transient, and reverses in a few years. Idiopathic juvenile osteoporosis is an uncommon self-limiting disease, manifesting in childhood, about 2 years before puberty, with inflammatory arthritis-type symptoms. In the appendicular skeleton, lucent lines in the metaphyses of long bones are noted, of unknown etiology, possibly due to defective osteoblastic function or increased osteoclastic activity, leading to excessive resorption of bone.7
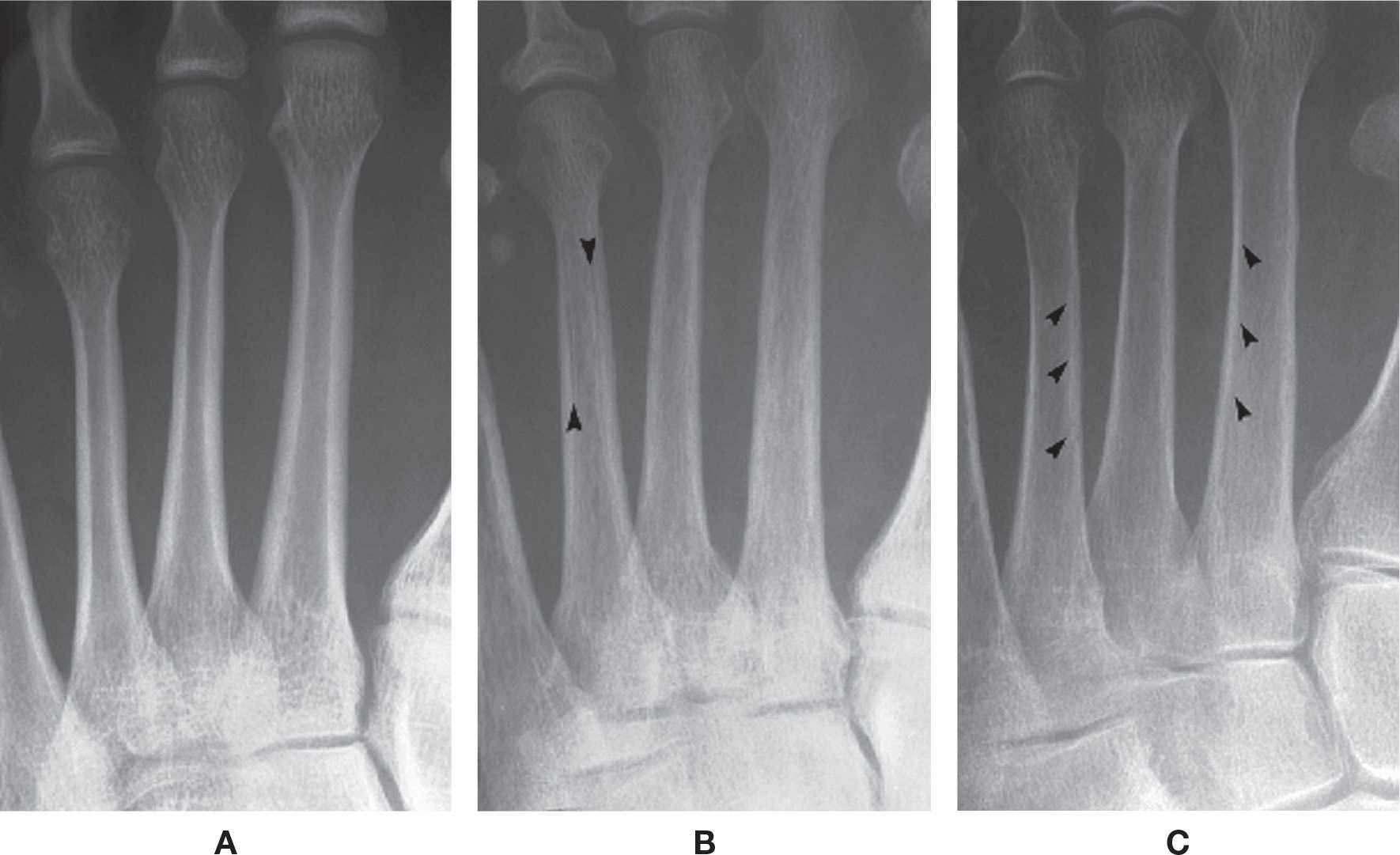
FIGURE 21-1. Chronic osteopenia, cortical bone. A: Normally, the endosteal and subperiosteal surfaces of the cortex are well-defined and continuous, and the remainder of the cortex is radiopaque and homogeneous in density. B: In chronic cortical osteopenia, the endosteal surfaces are ill-defined, and lucent striations (intracortical tunneling) may be seen running through the cortex (arrows), parallel to the shaft. C: Endosteal resorption (arrows), resulting in cortical thinning.
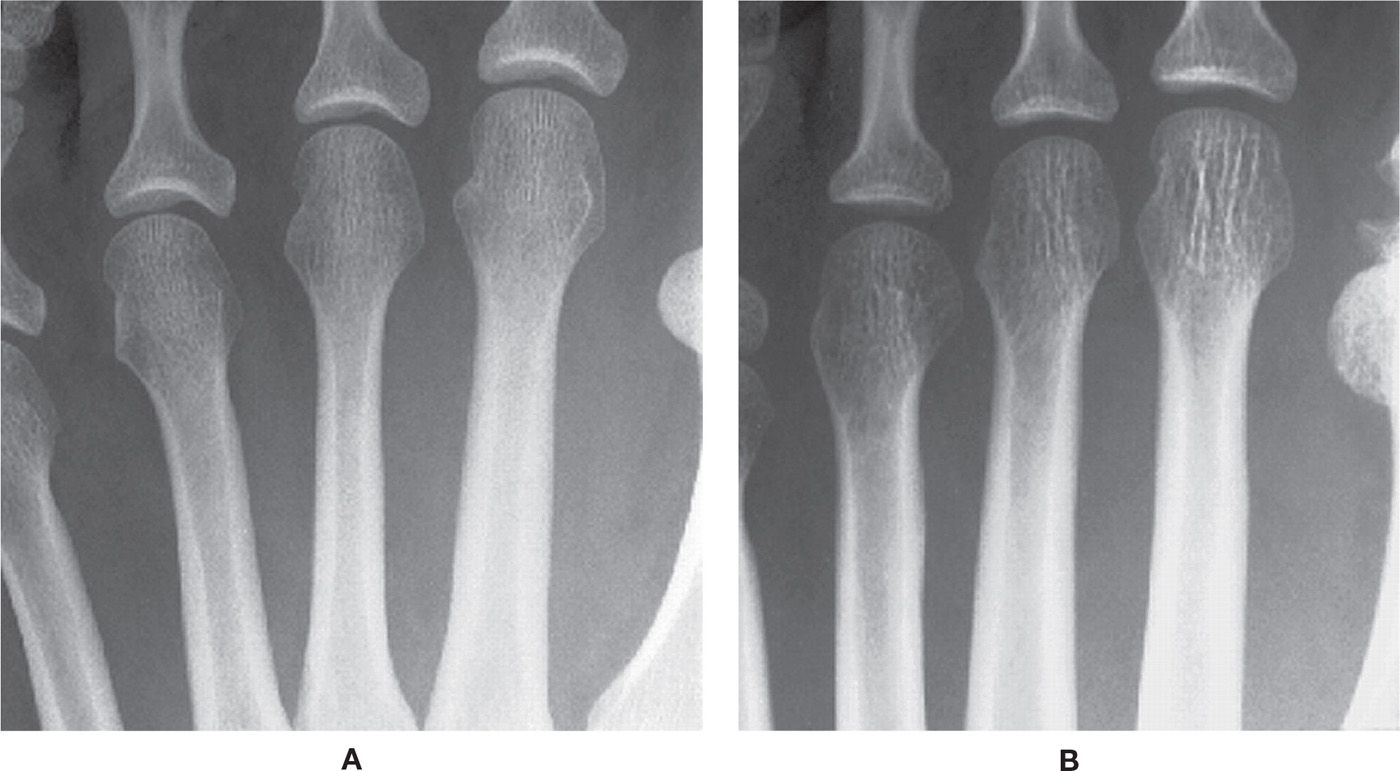
FIGURE 21-2. Chronic osteopenia, cancellous bone. A: Normal. Cancellous bone is made of primary and secondary trabeculae. The primary trabeculae—also known as stress trabeculae—are found along lines of stress. Secondary trabeculae are found perpendicular or oblique to the primary or stress trabeculae. Secondary trabeculae give spongiosa a fine, homogeneous appearance. B: With chronic osteopenia, secondary trabeculae are resorbed, leaving the primary trabeculae to stand out in relief. As bone is laid down on the remaining primary trabeculae, they become coarse in appearance.
Quantitative methods are necessary to assess bone mineral density and detect early osteoporosis; studies include single-energy x-ray absorptiometry (wrist and heel), radiographic absorptiometry (hand), single-photon absorptiometry (wrist), quantitative computed tomography (spine), and dual-photon x-ray absorptiometry (DXA; spine, hip, body), the gold standard. The metacarpal cortical index (MCI) has also been used to objectively measure osteoporosis. The thickness of the medial and lateral cortices of the second metacarpal shaft at its thickest point is measured; the sum of these thicknesses is divided by the total diameter of the shaft at that same level. The metacarpal cortical index is obtained by multiplying this fraction by 100.11 It has not been validated whether or not this index can be applied to the second metatarsal, but is has been used at this location for general evaluation.
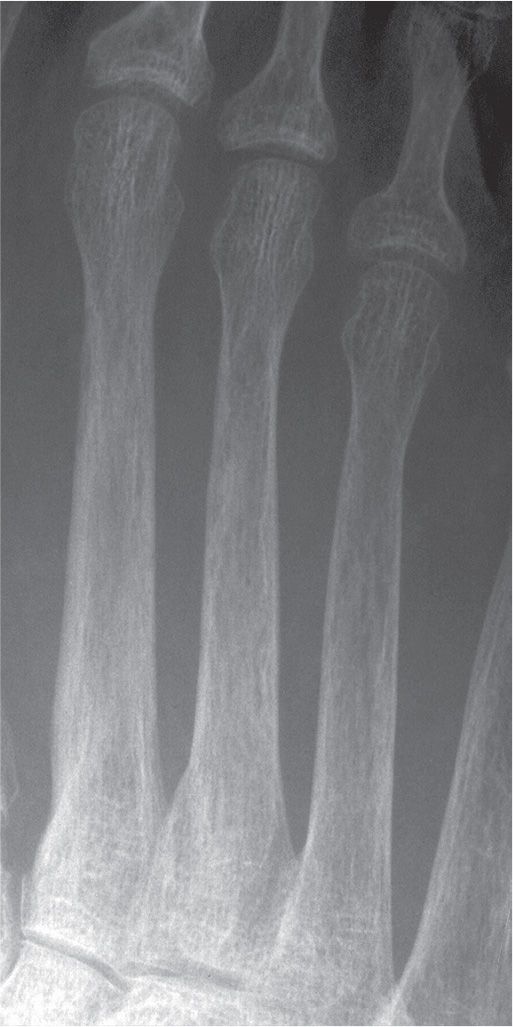
FIGURE 21-3. Generalized osteoporosis. This elderly, postmenopausal patient demonstrates the characteristic features of chronic osteopenia, including endosteal resorption, intracortical tunneling, and prominent, coarse primary trabeculae.
In addition to bone mass, trabecular bone architecture is important in assessing bone fragility, which is crucial in the diagnosis of osteoporosis. Radiography is a noninvasive two-dimensional technique used for this purpose in the appendicular skeleton, with good results, but it only reflects trabecular bone structure to a certain extent. It had been suggested that the appearance of calcaneal trabecular patterns provides an index for assessing osteoporosis12 and controversy existed as to whether or not these trabecular patterns correlate with actual bone density.13 However, Phan et al.14 cite recent studies that have established the importance of trabecular bone architecture along with bone mineral density, especially for assessing fracture risk. High-resolution MRI and computed tomography (CT) have limited spatial resolution, but have the potential to image three-dimensional architecture of trabeculations.15
It is well known that osteoporosis is not easily diagnosed with radiography, because 30% to 50% of skeletal mass must be lost before osteopenia is visually apparent.16 But recently Newton et al.8 reported that osteoporosis is often unrecognized, even in the presence of radiologic evidence of fracture, especially if due to low-trauma injury or in the elderly; osteopenia or fracture noted in foot radiographs are predictive of low bone mass, and these patients should be evaluated via DXA scan. Clinical risk factors for fracture in patients with osteoporosis, validated in randomized controlled trials, include low bone mineral density (DXA at spine or hip), prior vertebral fracture, long-term glucocorticoid treatment, and age.17
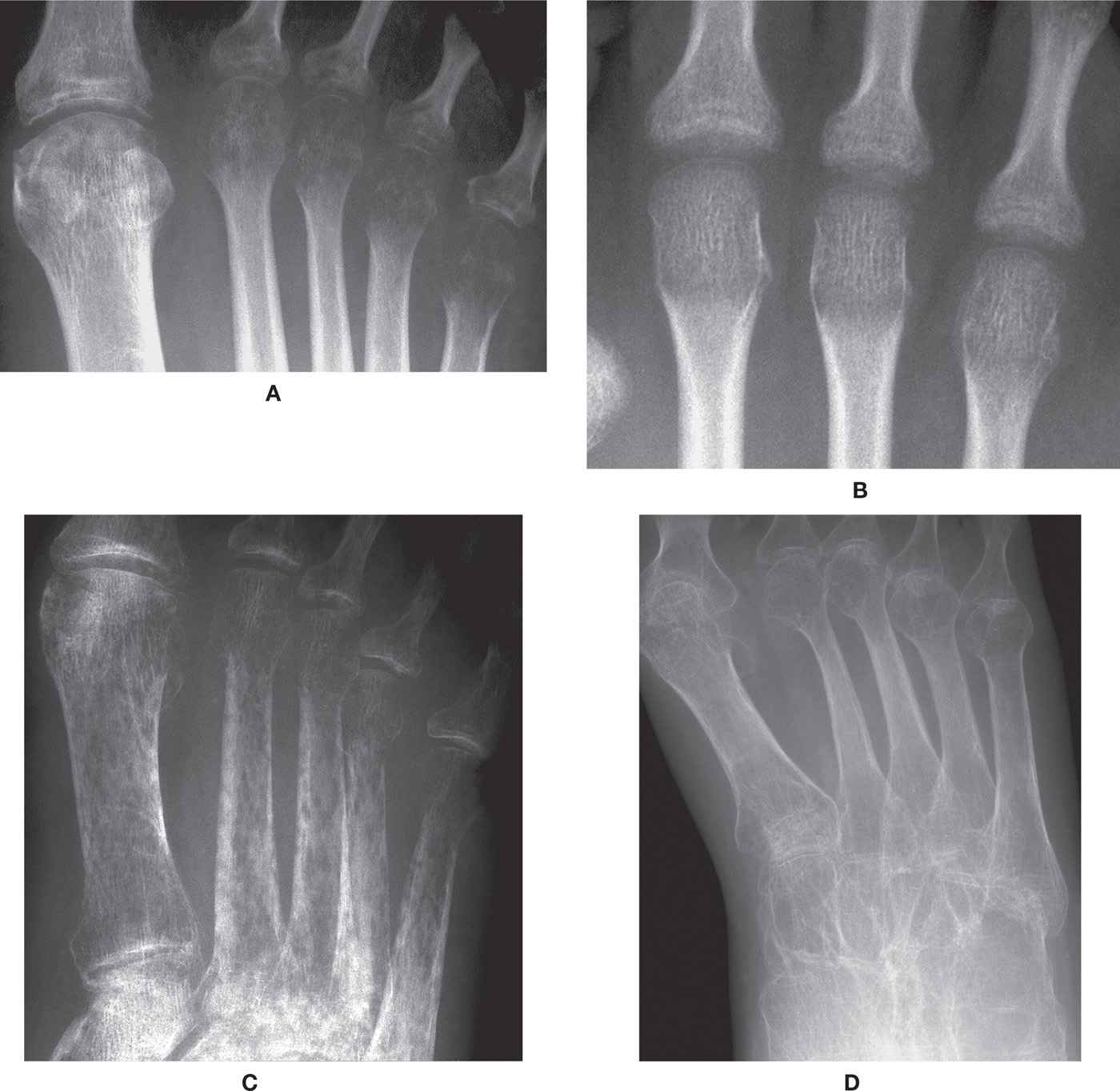
FIGURE 21-4. Regional osteoporosis. A: Periarticular, spotty (acute) osteopenia of cancellous bone at metatarsophalangeal joints. B: Metaphyseal ill-defined transverse bands of decreased density, all metatarsals (acute osteopenia). C: Severe acute osteopenia, with spotty, permeative decreased density of cancellous and cortical bone, and subperiosteal resorption. D: Severe osteopenia secondary to polio. Findings parallel those seen in chronic osteopenia (generalized osteoporosis).
Osteoporosis can also be subdivided based upon its distribution, as either generalized or regional.18 Generalized osteoporosis affects the entire skeleton; senile and postmenopausal osteoporosis are the most common. The mnemonic, ViNDICaTE, lists the differential diagnoses for generalized osteoporosis (Table 21-2). Senile osteoporosis simply refers to the gradual loss of skeletal mass that is seen with age. Postmenopausal osteoporosis refers to the increased bone loss seen in women following menopause. They both can commonly occur in the same individual, so the two entities are often lumped together.
The radiographic feature of generalized osteoporosis in the lower extremity is chronic osteopenia (Figures 21-1 to 21-3), which includes one or any combination of the following:
• Prominent primary trabeculations
• Thinning of the cortices
• Intracortical striations (tunneling)
| Differential Diagnoses for Generalized Osteoporosis (ViNDICaTE) |
Vascular | Anemia |
Nutritional | Scurvy, malnutrition, calcium deficiency |
Drugs | Steroids, heparin |
Idiopathic | — |
Congenital | Osteogenesis imperfecta |
Toxic | Alcoholism, chronic liver disease, cirrhosis |
Endocrine/Metabolic | Senile (aging), postmenopausal, pregnancy, diabetes mellitus, hyperparathyroidism, Cushing’s disease, acromegaly, hypogonadism |
Be aware that normal radiographic anatomy may be misinterpreted as chronic osteopenia. For example, the trabeculations in the first metatarsal head and neck are normally prominent and sometimes appear coarse.
Regional osteoporosis affects one extremity. It is a focal disorder limited to the affected limb instead of being a generalized process, and may be acute or chronic.18 It is associated with disuse (immobilization), complex regional pain syndrome (CRPS; previously known as reflex sympathetic dystrophy syndrome [RSD]), denervation, fracture, inflammation, and transient regional osteoporosis. Harris16 reported that bones near a fracture site, even if separated by a joint, show a high calcium uptake for months after the injury, and yet the bones are still osteopenic. The exact same findings were noted after immobilization or casting of a limb. The causes are controversial in the literature, but it seems that neural and vascular change in the area, as well as tonic muscle pull and weight-bearing stress, all may influence the remodeling changes.
The typical radiographic finding of regional osteoporosis is acute osteopenia (Figure 21-4) and will include one or any combination of the following:
• Spotty (moth-eaten, mottled, patchy) osteopenia in cancellous bone, especially periarticular regions, appearing as small, oval or round, lucencies within the affected bone
• Ill-defined broad (4–8 mm) transverse bands of decreased density at subchondral or metaphyseal locations
• Subperiosteal bone resorption (in severe cases, especially CRPS)
The radiographic appearance of acute osteopenia presents after 8 weeks of immobilization; in younger patients it appears earlier. The changes are reversible with return of activity.18 Table 21-3 summarizes imaging findings of CRPS.
Long-standing disuse causes uniform osteoporosis. The bone resorption starts at or just proximal to the fracture line and extends distally, involving the bones beyond the fracture.18 It affects most frequently the tarsal bones, as well as the bases of the metatarsals. Chronic disuse osteoporosis is often secondary to paralysis and it is associated with coarse, prominent primary trabeculations and cortical thinning (Figure 21-4D).
Transient osteoporosis, also known as regional migratory osteoporosis, has a predilection for pregnant women and middle-age men. It is a painful condition with no history of trauma that is uncommon and targets the lower extremity, primarily the hip and knee; it rarely affects the foot.19 Transient osteoporosis recovers spontaneously in 6 to 12 months, without late sequellae. Multiple joints of the same extremity may be involved. Radiographically, transient osteoporosis presents as severe osteopenia with relative preservation of the joint spaces, findings that are nonspecific. Bone scintigraphy and MRI have been used to further assess this disorder; however, they both show nonspecific findings (increased uptake and marrow edema, respectively).18
| Imaging Findings in Complex Regional Pain Syndrome18 |
Diagnostic Imaging | Findings |
Radiography | Spotty or patchy osteopenia in periarticular locations; when severe, may involve diaphysis as well |
Bone scintigraphy | Intense and diffuse isotope uptake in the affected bones in all three phases (flow, blood pool, and static imaging); sensitivity and specificity higher than 80% |
MRI | Normal or nonspecific soft tissue edema or marrow edema |
Osteomalacia
Osteomalacia (“softness of bone”) is characterized histologically as a disorder with excessive amounts of uncalcified osteoid.2 The inadequate or delayed mineralization of osteoid is a qualitative change in bone; in contrast, osteoid mineralization seen in osteoporosis is relatively normal except that it is decreased in quantity. Since osteoid formation exceeds matrix mineralization, total osteoid tissue is increased and appositional growth rate is decreased.16 Many metabolic disorders interfere with bone matrix deposition and mineralization and can cause osteomalacia. Etiologies are numerous and include vitamin D deficiency (dietary) and malabsorption (gastrointestinal disease), hypophosphatemia (oncogenic osteomalacia), and mineralization inhibitors (aluminum), to name a few.20
Osteomalacia is the given name when decreased bone mineralization is present in the adult skeleton; in the growing skeleton of infancy and childhood it is called rickets. In these conditions, the time lag between osteoid synthesis and mineralization becomes 2 to 3 months instead of the normal 5 to 10 days. Also, in the areas that do mineralize, persistence of large areas of decreased bone mineral density and low mineral content around osteocytes is noted.16
The radiographic features of osteomalacia are nonspecific, consisting primarily of osteopenia. Bowing deformity of long tubular bones may occur. A transversely oriented, incomplete radiolucency, known as pseudofracture (sometimes referred to as Looser zone and “milkman fracture”), may also be seen in tubular bones and is a classic radiographic sign.3 The lucent line may be bordered by sclerosis, is usually located along the compressive side of the bone, and tends to be bilateral and symmetrical. The femur is a common location for Looser zone.21
Hypophosphatasia
Hypophosphatasia is a condition characterized by reduced levels of alkaline phosphatase (ALP) in serum, bone, and other tissues, caused by a variety of mutations in the tissue nonspecific ALP gene. It is extremely rare, but has a frequency among Canadian Mennonite communities. Severity of the disease depends on the age of presentation; earlier ages have a more severe presentation; there is no curative treatment for the disease. Radiographically, characteristic abnormalities include bowing and shortening of long tubular bones as well as osteochondral spurring. There also may be rickets-like findings at the zone of provisional calcification.22 Chondrocalcinosis articularis and Looser zone are also seen. Looser zone tends to be on the outer cortex of long bone, thus helping in differentiating hypophosphatasia from osteomalacia, where the Looser zones are found to be on the inner cortex.21
Hyperparathyroidism
The general term hyperparathyroidism refers to increased levels of parathyroid hormone (PTH), which indirectly leads to increased osteoclastic activity and ultimately leads to the removal of calcium from bone that then enters the blood.3 Excessive PTH secretion has three forms. It may be due to parathyroid abnormality (such as tumor), which is referred to as primary hyperparathryroidism and leads to hypercalcemia. It may also occur in response to low calcium levels, as encountered in various situations such as vitamin D deficiency; this is referred to as secondary hyperparathyroidism. Tertiary hyperparathyroidism results from hyperplasia of the parathyroid glands and a loss of response to serum calcium levels; this disorder is most often seen in patients with chronic renal failure. In all cases, raised PTH levels are harmful to bone, and treatment is often necessary.
The characteristic radiographic feature of hyperparathyroidism is subperiosteal bone resorption (Figure 21-5). In fact, absence of this finding makes the diagnosis suspect. Other sites of bone resorption are periarticular, intracortical, endosteal, subchondral, and entheseal.
Histopathologically, fibrous tissue replaces the bone that is removed; this has been referred to as osteitis fibrosis cystica or Von Recklinghausen disease of bone.23 Numerous geographic radiolucent (lytic) lesions may be seen radiographically, corresponding to hot spots within the same bone on bone scan. These are referred to as brown tumors or osteoclastomas, which can resemble metastatic lesions; it is unusual to see them in the distal extremity, though a calcaneal location has been described recently.24
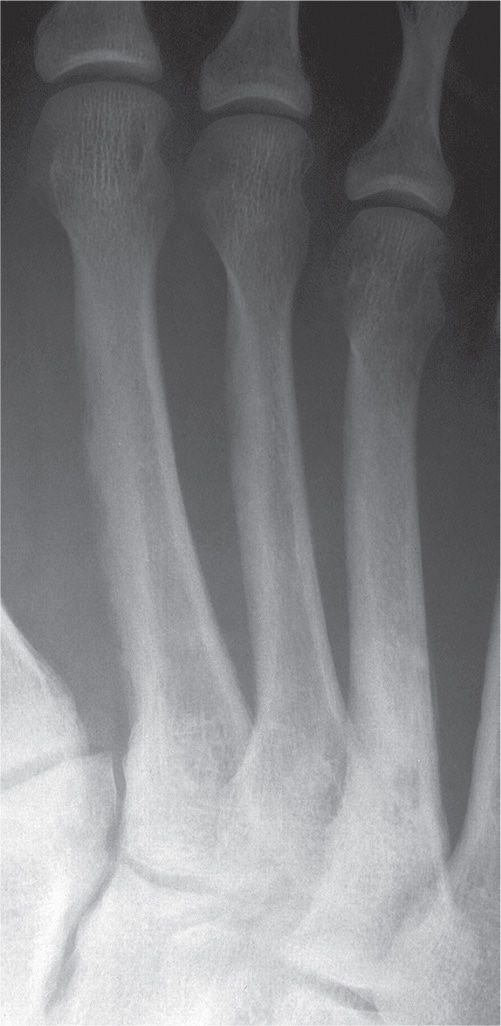
FIGURE 21-5. Hyperparathyroidism. Subperiosteal bone resorption along the medial aspects of the metatarsal shafts.
Primary hyperparathyroidism has been associated with hyperuricemia and overt gout; therefore, parathyroid adenoma should be considered with atypical presentations of gout (involving the ankle or talocalcaneal joints, for example).25 Soft tissue calcification may also be seen, especially associated with secondary hyperparathyroidism.
Renal Osteodystrophy
Patients with chronic renal failure demonstrate bony abnormalities known as renal osteodystrophy or renal bone disease.3 The symptoms in this disease are not usually seen in adults until they are on hemodialysis. Bone biopsies have shown both increased and decreased bone turnover. Chronic kidney disease causes hyperphosphatemia, which indirectly and directly results in an increase in PTH and stimulates osteoclast activity. Vitamin D activation and calcitriol activity also seem to play a role.20 Ultimately, renal osteodystrophy results from disturbances of calcium-regulating hormones and vitamin D and PTH metabolism, which cause disturbances in calcium-phosphate homeostasis.26 The radiographic findings manifested may parallel those of hyperparathyroidism, osteoporosis, and osteomalacia (or rickets if a child). Calcification of soft tissue and vessels is a frequent finding, especially when there are high serum calcium and phosphorus concentrations (Figure 21-6).
EPIPHYSEAL REGION ABNORMALITY
Rickets
Rickets is a disease of growing bone that is unique to children and adolescents. It is caused by a failure of osteoid to calcify in a growing person.10 Rickets involves impaired mineralization at the physis, resulting in deformity and impaired linear growth of long bones.21,27
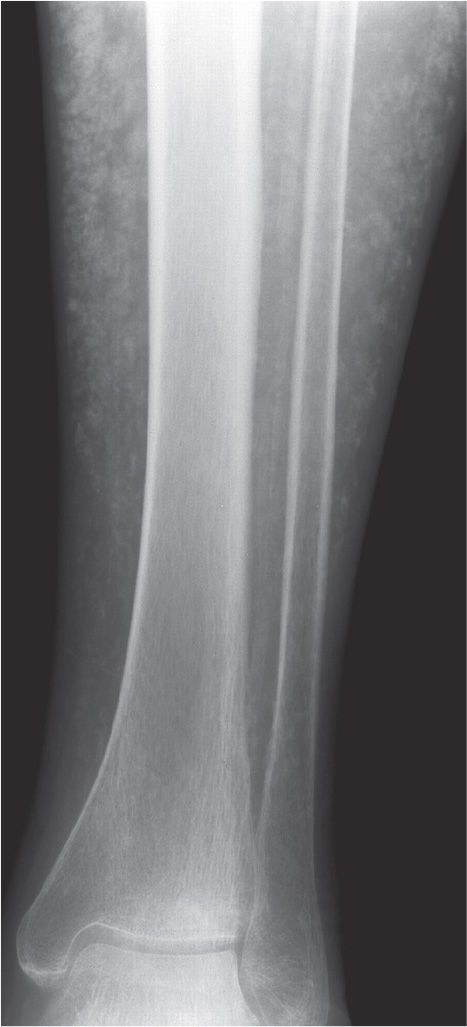
FIGURE 21-6. Renal osteodystrophy. Soft tissue calcification throughout the leg.
Rickets may result from vitamin D deficiency, abnormal metabolism of vitamin D, dietary calcium deficiency, or chronic hypophosphatemia.27 Several gastrointestinal disorders have been associated with decreased absorption of vitamin D, calcium, and phosphorus. Acquired or inherited renal tubular abnormalities that cause resorptive defects may result in rickets.10
Histologic changes are seen at the level of the growth plate or, more specifically, the zone of provisional calcification, where an increased number of disorganized cells are found. The increased number of cells results in increased width and thickness of the zone of provisional calcification.28
The radiographic features of rickets consist of both nonspecific and characteristic findings (Figure 21-7). Nonspecific findings include general retardation of body growth, osteopenia, and bowing deformity of long tubular bones. Characteristic changes occur at the growth plate region of tubular bones, where rapid bone turnover occurs (especially the tibial metaphyses). Findings include widening of the physis (due to excessive cartilage build-up), decreased density at the zone of provisional calcification (due to decreasing mineralization), irregularity of the physeal margin of the zone of provisional calcification, which has been described as “fraying” and a “paint-brush appearance” (due to variable calcification of the enlarged cartilage mass), and widening and cupping of the metaphysis (as the disorganized mass of cartilage expands).21,28 These findings are pronounced at the knee and ankle. In faster growing long bones, such as the tibia, transverse metaphyseal lines of increased density may be seen, representing the growth arrest and recovery lines of Harris or Park.7,29 Genu varum or genu valgum may be seen in crawling toddlers.21
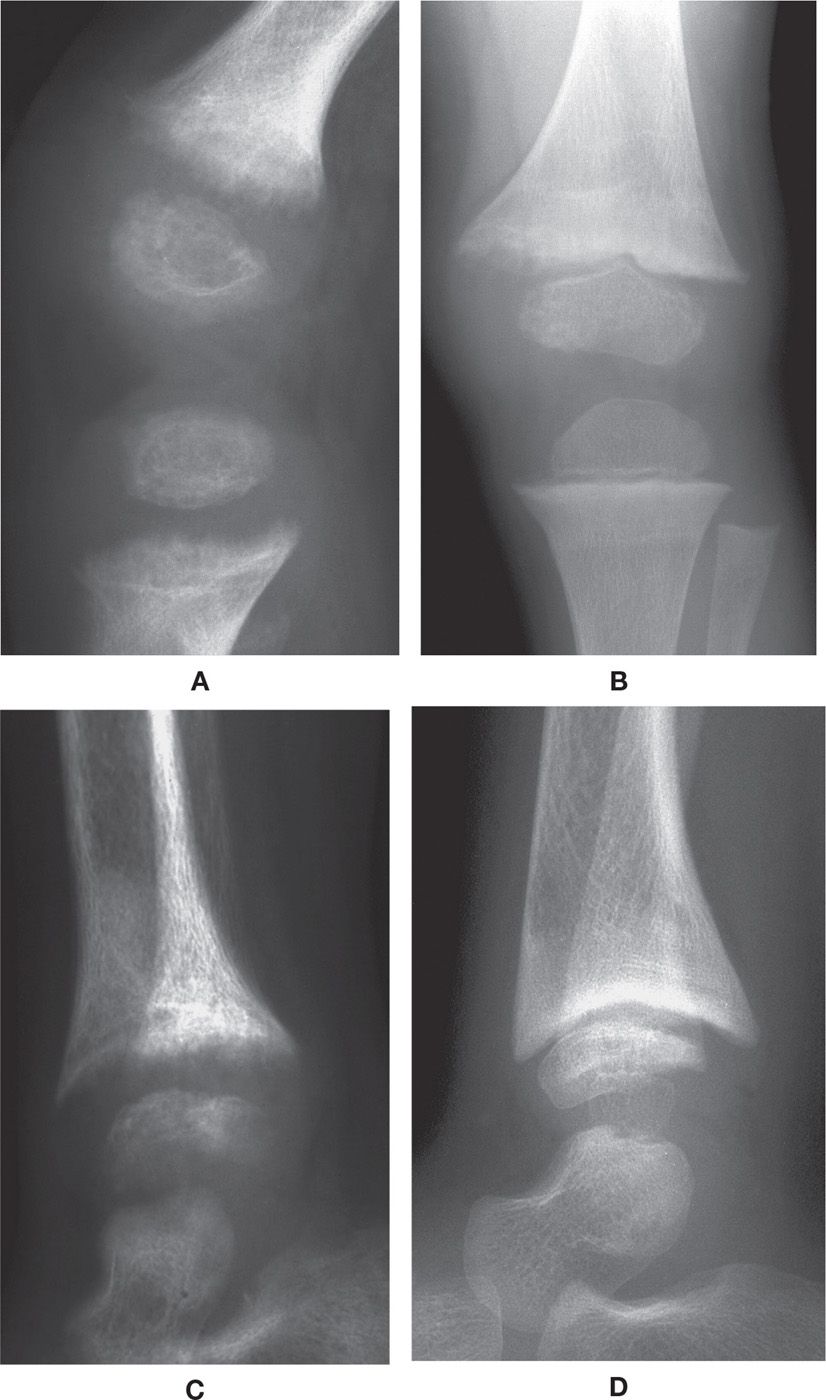
FIGURE 21-7. Rickets. A: Knee, lateral view. Characteristic features include an ill-defined, lucent and frayed zone of provisional calcification, with widening of the physis. B: Knee, anteroposterior (AP) view. Following treatment, the zone of provisional calcification has become more defined, although still irregular and ill-defined medially. C: Ankle, lateral view. Same date as A; similar findings demonstrated, with additional metaphyseal cupping. D: Ankle, lateral view. Metaphyseal cupping very prominent following treatment; also note defect along surface of talar dome.
Scurvy (Hypovitaminosis C)
Scurvy is caused by insufficient dietary intake of vitamin C, which is required for the synthesis of collagen that is an important part of bone.30 There are two forms: pediatric scurvy (Barlow disease) and adult scurvy. Unfortunately, it is still common today, primarily due to lack of fresh fruit and vegetable in the diet. Risk factors include bottle-fed infants with no vitamin C supplementation, the elderly, males living alone, chronic alcoholism, and high cigarette usage. Clinical manifestations are related to fragility of vessel walls leading to bleeding; damage to synovial blood vessels and microfractures cause bleeding in joints.31
The characteristic radiographic features (Figure 21-8) occur in the developing child. In the metaphyseal region they include: (1) a transverse line of increased density bordering the growth plate, due to the thickened zone of provisional calcification (the “white line of scurvy/Frankel”); (2) a transverse line of decreased density adjacent to the line of increased density on its metaphyseal side that consists of detritus (referred to as the “scurvy line,” scorbutic zone and Trümmerfeldzone); (3) small, beak-like outgrowths or extensions of the zone of provisional calcification along its margins (“Pelkan spur/beak”); and (4) subepiphyseal infractions in an area of brittle and decreased trabeculae (“corner sign,” “angle sign”).3,32 The epiphysis appears as an outer shell of increased density surrounding a central lucency, due to atrophy of the central spongiosa (also known as “ring epiphysis” and Wimberger sign). Extensive periostitis may be seen along the entire length of the bone secondary to subperiosteal hemorrhage stimulating the periosteum.3
Radiographic findings are limited in the adult, despite the extremity pain and swelling. There may be osteoporosis and, rarely, due to hemarthrosis, bone resorption at joints.31
ALTERED BONE FORM
Acromegaly
Growth hormone (GH) is fundamental for the maintenance of bone mass and metabolism during childhood, as well as in the adult. Osteoblast proliferation is regulated by GH and insulin-like growth factor type I (IGF-I). IGF-I overexpression increases bone mineral density with trabecular bone volume, while GH overexpression leads initially to increased bone formation, followed by increased bone resorption. Overall, GH and IGF-I excesses increase the cortical bone density, regardless of age or gonadal function.33,34
A number of disorders may increase GH output; most commonly it is related to a pituitary adenoma. In the adult, this disorder is known as acromegaly, and can result in severe disfigurement, serious complicating conditions, and premature death if unchecked; gigantism results in the child with open growth plates. Acromegaly causes increased bone turnover and appendicular cortical bone mass. The rate of fracture in acromegalic patients is low despite the high prevalence of osteoporosis. This seems to be related to the anabolic effect of GH on trabecular and cortical bone, which appears to persist even after remission of acromegaly.33
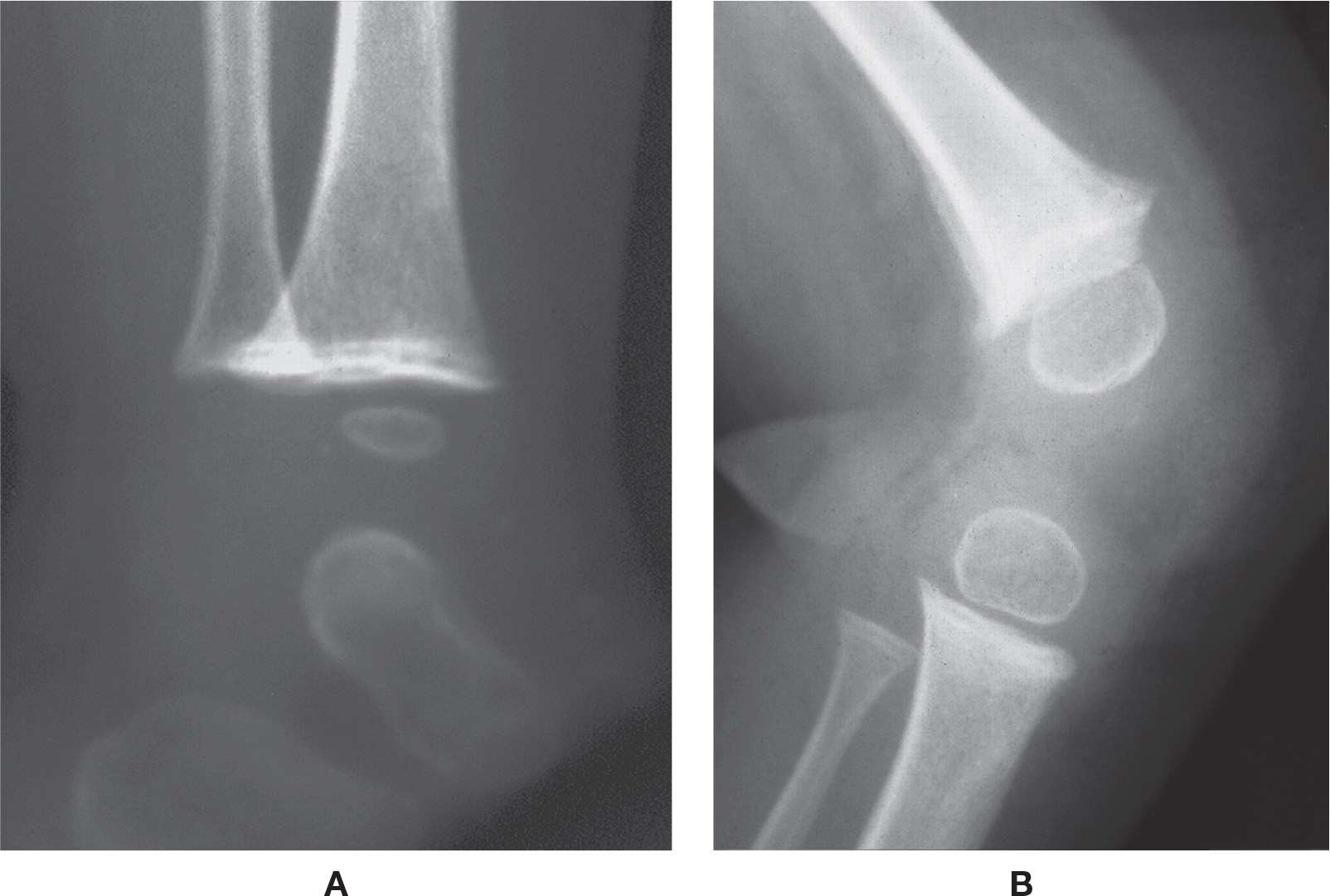
FIGURE 21-8. Scurvy. A: Ankle. Note the transverse line of increased density along the metaphysis. B: Knee (different patient). In addition to the sclerotic line across the metaphysis, beaks are visible at the metaphyseal margins, and Wimberger’s sign is evident.
Radiographically, the patient with acromegaly demonstrates abnormalities of soft tissue, joint space, and bone (Figure 21-9).2 The increase in soft tissue volume is reflected by a heel pad thickness that is greater than 25 mm in the male and 23 mm in the female (when local causes are excluded).3 The joint spaces appear widened due to cartilage thickening, which is caused by GH and IGF-I induced replication of articular chondrocytes and increased matrix synthesis.33 Bones become prominent; the metatarsal heads and distal phalanx ungual tuberosities are enlarged, metatarsal shafts are thickened, and spurs are found at entheses. Interestingly, however, the proximal phalangeal shafts may appear narrow in girth.
Acromegalic arthropathy, which resembles osteoarthritis, may occur; radiographic findings consist of osteophytes, eburnation, subchondral geode formation, joint space narrowing, and calcification of ligament and joint capsule entheses. The literature shows that up to 75% of patients with acromegalic arthropathy have only mild manifestations, while approximately 33% present with severe arthropathy. Arthropathy affects most acromegalic patients, and is the leading cause of morbidity and functional disability. Radiologic signs can be detected early in the course of the disease and usually precede other clinical manifestations, which is important because only the early stages seem to be reversible by effective treatment. Ultrasonography is valuable in evaluation of joint capsule and periarticular tendons, and it shows increased joint thickness in acromegalic patients.33
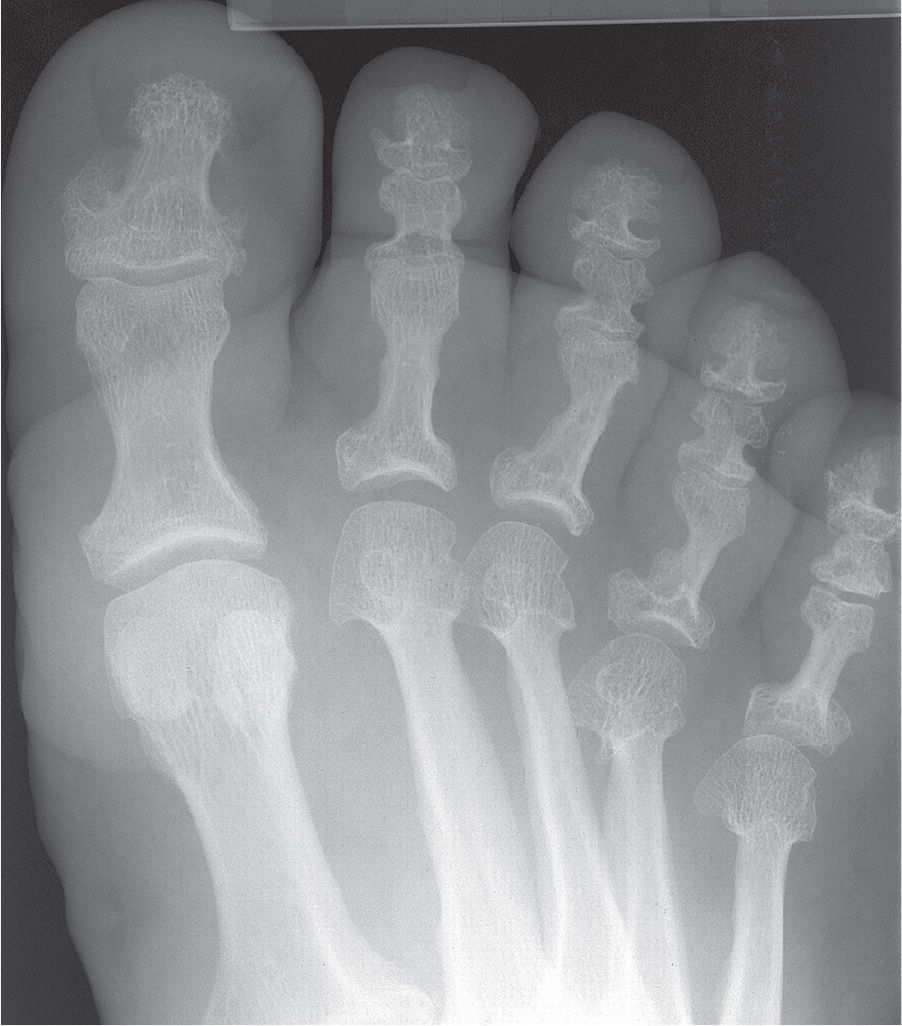
FIGURE 21-9. Acromegaly. Characteristic features include enlargement of bones (especially metatarsal heads and shafts and phalangeal bases) and soft tissue thickening (increased soft tissue density and volume).
Hereditary Multiple Exostoses
Hereditary multiple exostoses or osteochondromatosis is a skeletal dysplasia resulting from a disturbance of chondroid production resulting in heterotopic proliferation of epiphyseal chondroblasts.35 This relatively uncommon disorder of enchondral bone (although the most common benign bone lesion) has been known under different names as well, including osteochondromata, hereditary multiple osteochondromas, hereditary cartilaginous exostoses, hereditary deforming dyschondroplasia, and diaphyseal aclasia.36
Hereditary multiple exostoses is an autosomal dominant disorder. The exostoses are usually found bilaterally and symmetrically; however, the number of them varies widely. In the lower extremity, they target the knee and ankle, though they are found also in the metatarsal and phalanx; it rarely affects the calcaneus. Each individual exostosis appears identical to the benign bone tumor osteochondroma, which is a cartilage-capped exostosis adjacent to the diaphyseal side of the physis. It appears in the first two decades of life as painless bumps near the ends of long bones (usually the tibia and femur).36,37
Radiographically, the exostosis is found adjacent to the metaphysis; it may be attached to the bone by a stalk or pedicle (pedunculated) or lack a stalk (sessile) (Figure 21-10). When pedunculated, the exostosis faces away from the nearest physis; the sessile exostosis is broadly attached. The cortex and cancellous portions of the exostosis are contiguous with the parental bone. It can cause bone deformity (ankle and genu valgum) and shortening (short stature; limb length discrepancy; brachymetatarsia). A large periarticular exostosis can limit a joint’s range of motion. Approximately 5% of these lesions transform into chondrosarcoma.36,37
Enchondromatosis
Enchondromatosis (Ollier disease) may mimic osteochondromatosis.38 However, the abnormality is due to multiple benign cartilage tumors (enchondromas) within bone as opposed to being along the outer surface of bones. They frequent the phalanges and the tibia in the lower extremity, and are usually asymmetrical with unilateral predominance. Also, enchondromatosis is nonhereditary and appears spontaneously. They cause greater bone deformity than do multiple hereditary exostoses. The risk of malignant transformation (into chondrosarcoma) has been reported from 20% to 50%. Maffucci syndrome is the name given to multiple enchondromas associated with hemangiomas; its prognosis is more severe than enchondromatosis.37
Radiographically, the enchondroma is a geographic destructive (lucent) lesion that is located centrally in a bone. The cartilage matrix may calcify, and appear punctate throughout the lesion. Bone deformity and shortening is greater than that seen in multiple hereditary exostoses.37
Osteogenesis Imperfecta
Osteogenesis imperfecta is a heterogeneous group of inherited disorders characterized by an involvement of bones and extraskeletal tissues (skin, teeth, sclerae, and ligaments). It is the result of abnormal metaphyseal and periosteal ossification caused by deficient osteoid production.35 It is sometimes referred to as brittle bone disease, with increased tendency to fracture.39 Abnormal maturation of collagen occurs in both mineralized and nonmineralized tissues.
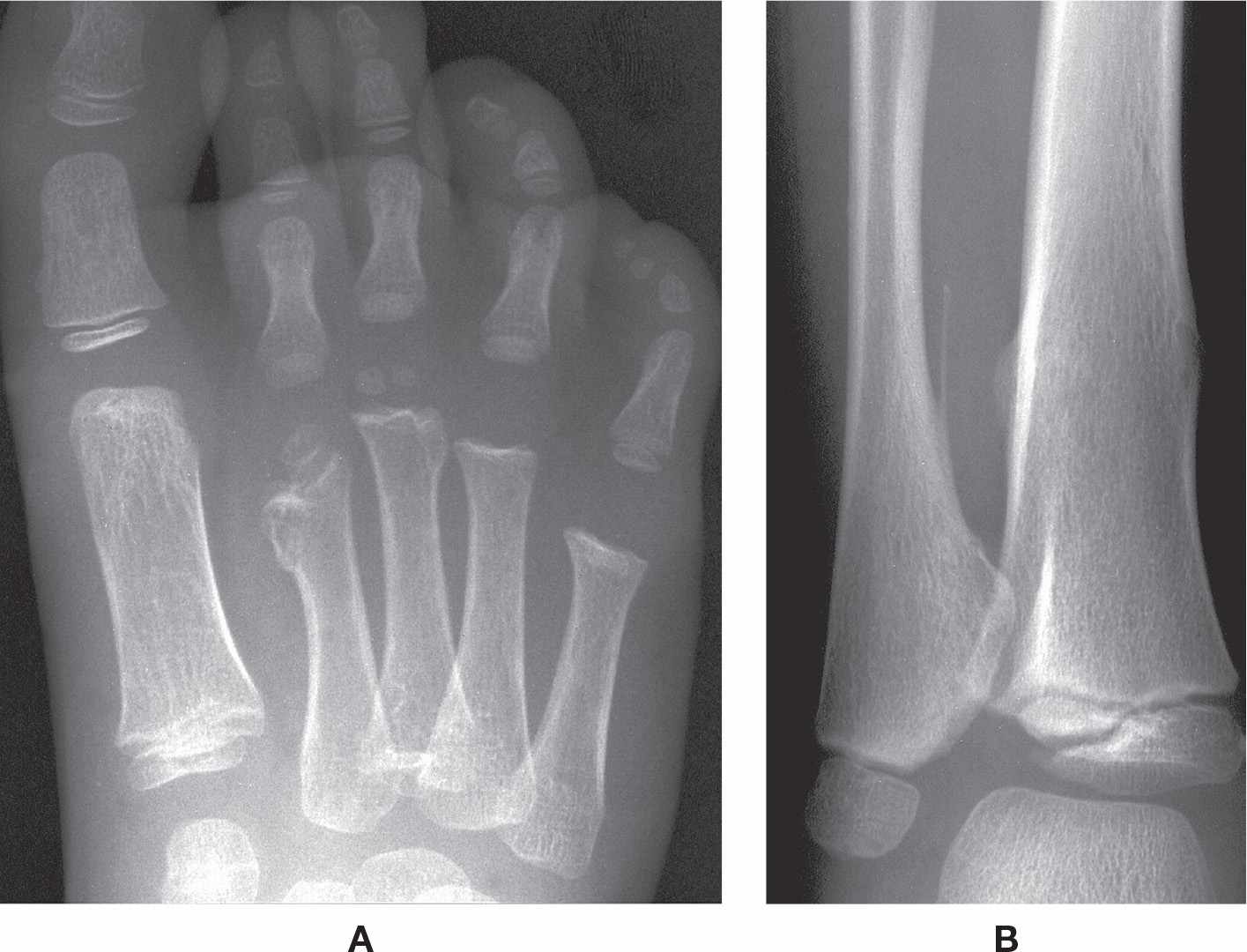
FIGURE 21-10. Hereditary multiple exostoses. A: DP view. Sessile exostoses of the second and third metatarsals, with shortening of the second. B: Same patient, AP view, ankle. Exostoses are seen along the medial aspect of the distal fibular diaphysis (pedunculated) and the medial and lateral aspects of the tibial diaphysis (sessile).
There are four different types of osteogenesis imperfecta. (Five additional types, V–IX, are variants of these.) Type I is most common, a mild form that demonstrates fractures but no dwarfing or bone deformity; type II is neonatal and lethal; type III is rare and demonstrates dwarfing and extremely fragile bones (fractures are common); and type IV has variable findings ranging from marked short stature and bone deformity to normal height.3 The tendency to fracture in types I and IV lessens in adolescence.20
Radiographic features more commonly include diffuse osteopenia (presenting occasionally with coarse trabeculae), diminished bone girth, and flared metaphyses (Figure 21-11). An obvious complication is fracture, which may demonstrate exuberant bone callus formation.
Hypoparathyroidism and Albright Hereditary Osteodystrophy
Hypoparathyroidism is caused by deficient or absent PTH. Its etiology is diverse, but is most commonly due to surgical excision of all parathyroid tissue (iatrogenic hypothyroidism); another important cause is an autoimmune disorder involving antibodies that activate the calcium-sensing receptor of the parathyroid glands (idiopathic hypothyroidism).40,41 Independent of etiology, hypoparathyroidism is a PTH-deficient or -resistant state characterized by hypocalcemia, hyperphosphatemia, and hypercalciuria.42,43 Radiographic findings of hypoparathyroidism in the lower extremity are limited. There may be osteosclerosis of bones and soft tissue calcification.
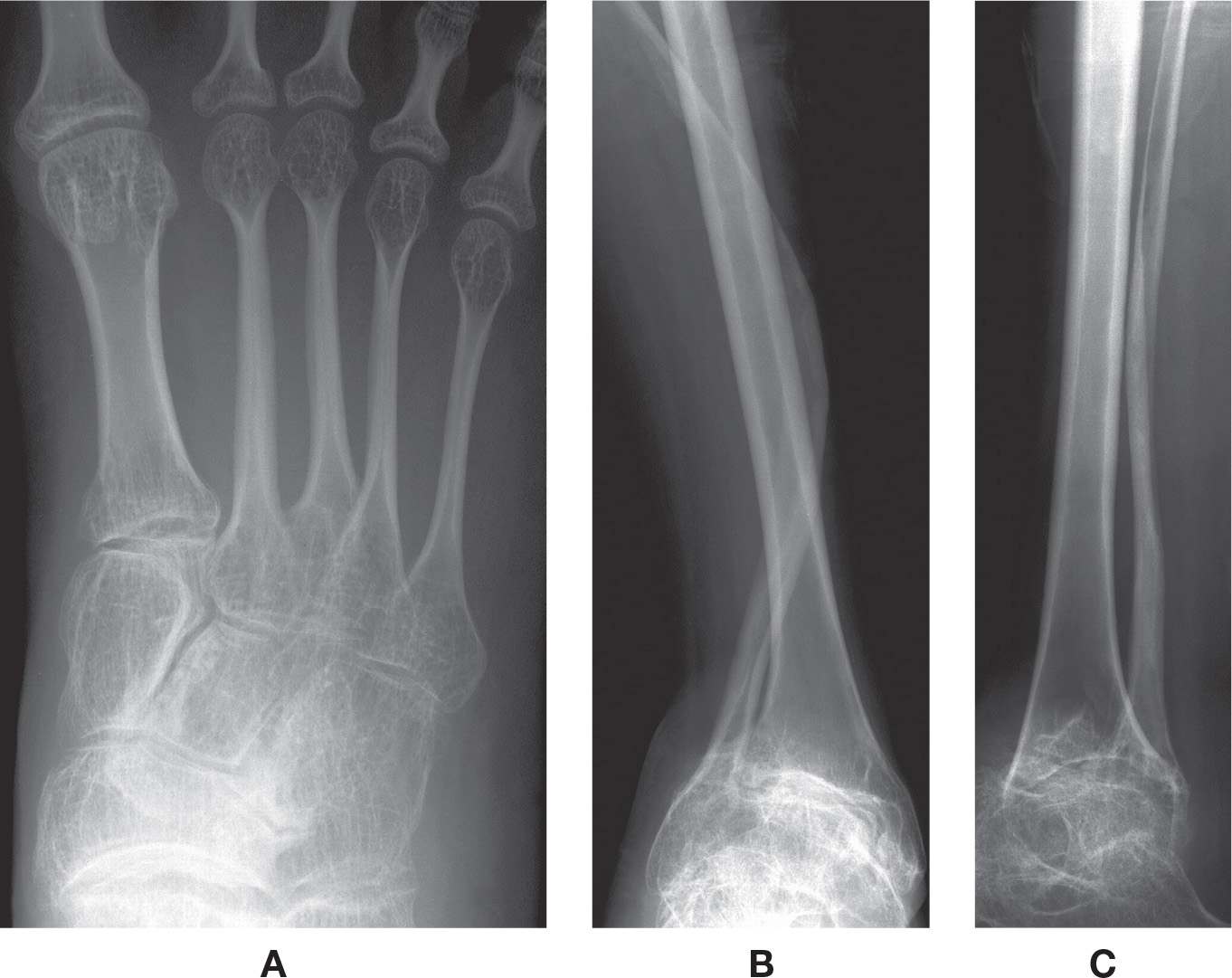
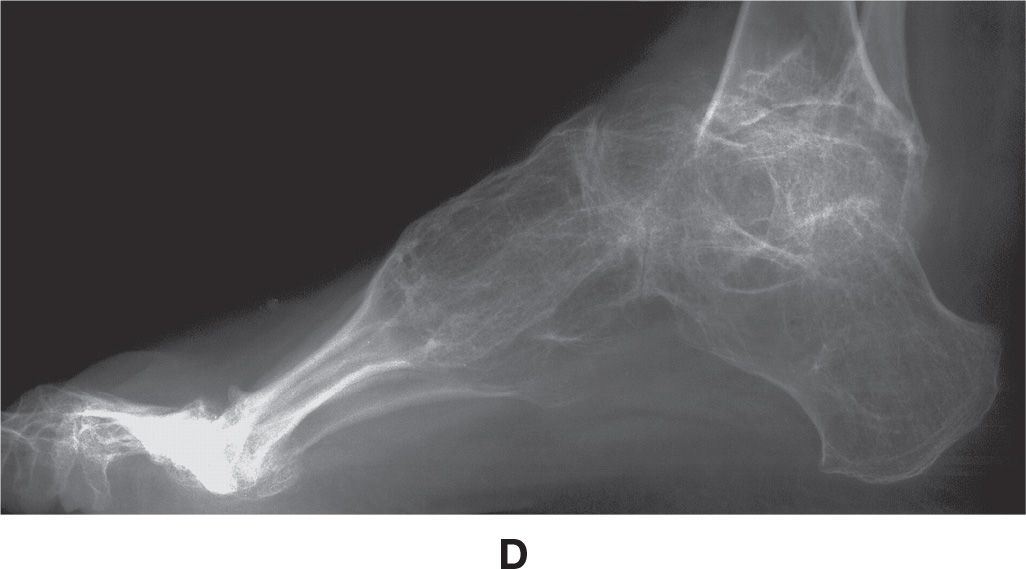
FIGURE 21-11. Osteogenesis imperfecta. A: DP view, foot. Characteristic features include narrowed girth of tubular bones and cancellous osteopenia. B–D: Different patient demonstrating severe osteoporosis and severe deformity of the fibula.
Rarely, there is tissue resistance to the actions of PTH, which results in a picture of hypoparathyroidism but with elevated PTH levels; this is known as pseudohypoparathyroidism.41 Pseudohypoparathyroidism is due to a genetic disturbance, confirmed by its occurrence in twins, as well as in mother and child. The genetic defects may cause effects exactly similar to those of true hypoparathyroidism, except that the injection of PTH fails to increase phosphaturia, indicating abnormal peripheral response to PTH; parathyroid gland biopsy shows them to be normal or hyperplastic.44
In 1942, Fuller Albright45 first introduced the term pseudohypoparathyroidism to describe patients who presented with PTH-resistant hypocalcemia and hyperphosphatemia along with an unusual constellation of developmental and skeletal defects, which have been collectively termed Albright hereditary osteodystrophy (AHO). These features include short stature, rounded face, brachydactyly, obesity, dental hypoplasia, and soft tissue calcification/ossification.44 Radiographic features include short metatarsals and phalanges, exostoses, and widening of bones. Brachymetaphalangia (shortening of tubular bones) is widely associated with AHO, though it is not pathognomonic.46 AHO has also been associated with a radiographic developmental variant, the cone-shaped epiphysis.3
Pseudopseudohypoparathyroidism is an inherited disorder as well and is named for its similarity to pseudohypoparathyroidism in presentation. The term pseudopseudohypoparathyroidism is used to describe a condition where the individual has the phenotypic appearance of pseudohypoparathyroidism, but is biochemically normal, that is, the calcium levels are normal.3 Both pseudohypoparathyroidism and pseudopseudohypoparathyroidism are considered forms of AHO.
ALTERED BONE ARCHITECTURE
Paget Disease
Paget disease (osteitis deformans) is the second most common aging bone disorder, following osteoporosis (and excluding fracture).47 It is a chronic disorder characterized by excessive and abnormal remodeling of bone, resulting in enlarged and deformed bones (Figure 21-12).3 It affects 3% to 4% of those who are older than 40 years, occurs slightly more frequently in men, and has a prevalence of 10% in the population over 70 years of age.48 Its etiology is still unknown, with controversial evidence; both genetic and environmental factors have been implied, as well as viral.49 It has been associated with marked elevation of serum ALP and hydroxyproline in serum and urine.3 It may or may not be symptomatic.
Paget disease can affect any bone but predominates in the axial skeleton. The most common tubular bones affected in the lower extremity are the femur and tibia; it seldom involves the calcaneus.47 Paget disease can either be monostotic (10%–35% of cases), or polyostotic (65%–90% of cases).3 It is typically asymptomatic in the foot, which may explain the infrequent reports at this location in the literature.47,50
Paget disease evolves through three stages of pathologic activity that represent a continuum. The first is the incipient and active lytic phase, which is characterized by overactive giant multinucleated osteoclasts that resorb bone. This is followed by the active mixed phase where both osteoblasts and osteoclasts are present; excessive osteoblastic activity is disordered, laying down architecturally abnormal bone at the affected sites.49 The late, inactive blastic phase, also referred to as the silent or quiescent stage, results as osteoblastic activity declines.3,48
The three pathologic stages of Paget disease have been confirmed radiographically. The first stage is described as osteolytic, affecting especially the skull (osteoporosis circumscripta) and long bones; it is often short-lived.48,49 Resorption starts in the subchondral area, spreading to the metaphysis and eventually the diaphysis; a lucent wedge results in what has been described as a “blade of grass” or “flame” appearance, estimated to progress at a rate of 1 cm per year.51 The second stage is mixed and demonstrates both osteolysis and osteosclerosis. The diaphysis appears lucent, whereas the epiphyseal and metaphyseal regions are sclerotic. In the long bones sclerosis may be extensive, obliterating areas of previous trabecular thickening. Bone in the third stage is predominantly sclerotic, and involves mainly the proximal long bones. Findings include cortical thickening, bone enlargement, and coarsened trabeculae. If the tibia is affected, there may be anterior bowing, which has been referred to as “sabre-shin deformity”; pseudofracture (similar to that seen in osteomalacia) may be seen along its anterior (convex) surface as well.23,52
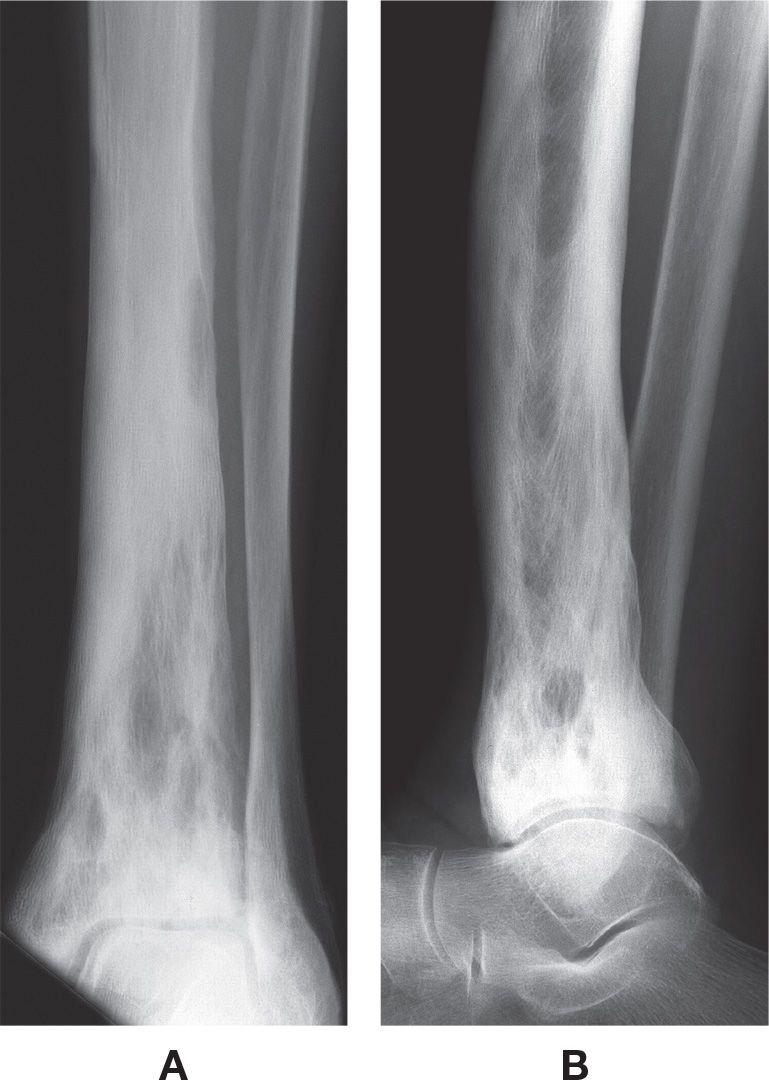
FIGURE 21-12. Paget disease. A: Mortise view. B: Lateral view. The bone is predominantly sclerotic in the diaphysis and metaphysis, with mixed well-defined lucent areas intermixed. Probably the third stage.
Bone scintigraphy, with its advantage of surveying the entire skeleton, demonstrates increased uptake of radionuclide in the region of abnormal bone in all phases of Paget disease.47 Scintigraphy is sensitive but not specific for detection of hyperemia and osteoblastic activity seen in Paget disease. The area of abnormal uptake is usually elongated, thus helping in differentiating Paget disease from metastatic diseases or myeloma, in which the radionuclide uptake is circular. Since bone scintigraphy is sensitive in detecting hypervascularity, a marked uptake of radionuclide is noted even before the typical radiolucency becomes evident on radiographic imaging, and is very valuable in detecting the polyostotic form of the disease. However, in the late, quiescent, phase of the disease radionuclide uptake may be normal, while the radiographs are abnormal. It has been noted that patients with a normal appearing bone scan and abnormal radiographic findings were asymptomatic, while those with abnormal bone scans and normal radiographs were symptomatic, with more active bone disease.48
CT and MRI usually show changes similar to radiographic imaging in noncomplicated Paget disease. The disorganized pattern of trabecular thickening is better demonstrated on CT than on radiographs. The bone enlargement, as well as cortical and trabecular thickening, pathognomonic signs of Paget disease, are seen with CT and MRI, but are better appreciated on radiographs.48
Complications associated with Paget disease include osteoarthritis, fracture, osteomyelitis, and neurologic complications.3,51 Metastasis of pagetic bone, sometimes considered a fourth phase, has been reported in 1% to 10% of patients.30 Sarcomatous transformation, occurring in about 1% of the cases, is the most feared complication. Although radiography is a more economical method for diagnosing Paget disease, MRI is more suited for demonstrating the presence and extent of neoplastic and nonneoplastic complications of this disease.53
Juvenile Paget Disease
Juvenile Paget disease, also called idiopathic hyperphosphatasia, is an abnormally high turnover bone disease, causing a significant increase of plasma ALP as well as increased levels of N-telopeptide and hydroxyproline (markers of bone resorption). Idiopathic hyperphosphatasia has an autosomal recessive inheritance.54
Radiographically, patients with severe phenotype show widened, osteopenic long bones with coarse trabeculae; cortical bone may be indistinguishable from the medullary canal. Patients with a milder phenotype demonstrate intense cortical thickening, especially along the medial aspect of the femur. A common finding is periostitis. The deformity in long bones is progressive in all phenotypes. Bone scintigraphy demonstrates diffuse increased uptake throughout the skeleton.54
Fibrous Dysplasia
Fibrous dysplasia is a benign condition where normal bone is replaced by fibro-osseous tissue. There is an inability of bone-forming tissue to produce mature lamellar bone from woven bone. It has also been associated with skin hyperpigmentation and endocrine dysfunction.55 Fibrous dysplasia is caused by somatic mutation of a gene that codes for a G-protein receptor.56 Pathologically, normal bone undergoing physiologic resorption is replaced with fibrous tissue.23 It may occur in one (monostotic, 75% of cases) or many (polyostotic, 25% of cases) bones in those between the ages of 10 and 30. In tubular bones its location is intramedullary and diaphyseal, with only occasional epiphyseal involvement.3 In the lower extremity it occurs more commonly in the tibia. It is associated with pain, limp, and leg-length discrepancy.56
Radiographically, the lesion is somewhat radiolucent with a hazy, ground-glass (homogeneous) quality, and may appear expansile with endosteal scalloping (Figure 21-13). Involvement of the femoral neck and adjacent shaft results in what is known as a “shepherd’s crook” deformity.56 There is rare potential for malignant transformation.
Anemia
Chronic bone marrow hyperplasia results in trabecular coarsening and cortical thinning.57 Complications are related to interruption of blood supply, which leads to infarction of bone.
Sickle cell disease is caused by production of abnormal hemoglobin, which binds with other abnormal hemoglobin molecules within the red blood cells; this causes rigid deformation of the cell (sickle shape), resulting in sludging and congestion of vascular beds, followed by ischemia and tissue infarction. An acute vaso-occlusive complication is commonly seen in black children with sickle cell disease that are at least 6 months old to less than 6 years of age. Known as dactylitis, this vaso-occlusive crisis is a painful swelling of the feet, toes, hands, and fingers, associated with fever.58
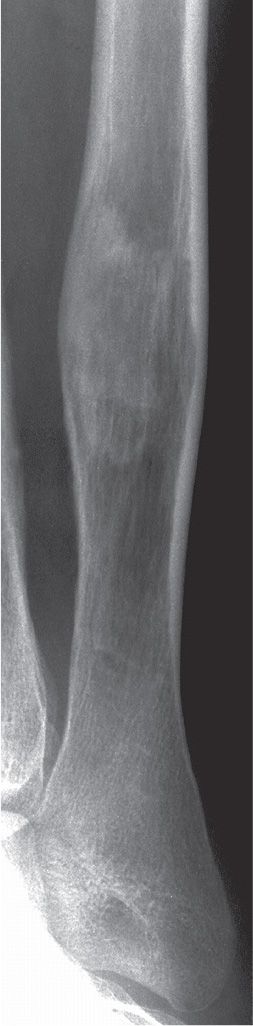
FIGURE 21-13. Fibrous dysplasia. The geographic lesion in the distal fibular diaphysis demonstrates a hazy, ground-glass appearance centrally. Notice its intramedullary location and cortical expansion with endosteal resorption.
Dactylitis, also known as the “hand-foot syndrome,” is often the first manifestation of sickle cell anemia and is frequently misdiagnosed. Histopathologically, it appears as extensive infarction of the marrow, medullary trabeculae, and the inner layers of cortical bone; there is also periosteal elevation and periostitis.58 Radiographically, the feet appear normal initially; within 10 days of the onset of symptoms, periostitis occurs first, causing the affected metatarsal or phalanx to appear rectangular in shape. Thinning of cortical bone and attenuation of the medullary canal follow periostitis, accompanied by irregular densities in the medullary canal.59
Radiographs obtained early during pain crises may be normal due to the fact that ischemia causes pain even before infarction is manifested. Patients with sickle cell anemia are also prone to silent infarction, and the discovery of osteonecrosis may be an incidental finding.58,60 (The radiographic features of epiphyseal osteonecrosis and bone infarct are described and illustrated in Chapter 17.)
Osteomyelitis is a serious complication of sickle cell anemia. It is most commonly seen in the diaphyseal region of the long bone (femur, tibia), and involves infection with Salmonella, followed by Staphylococcus aureus, and gram-negative enteric bacilli.
Thalassemia, also known as Cooley anemia, is a common hereditary disorder characterized by abnormal hemoglobin synthesis.23 There are three forms, major, minor, and intermedia; thalassemia major is the most severe and demonstrates the most common radiographic features.
Radiographic findings of thalassemia mirror the degree of marrow hyperplasia and include widening of the marrow cavity, thinning of the cortex, and coarse trabeculations that are sometimes described as having a “honeycomb” appearance (Figure 21-14).3,23 Widening of the metaphysis and epiphysis of a long tubular bone resembles the appearance of an “Erlenmeyer flask.”3,23
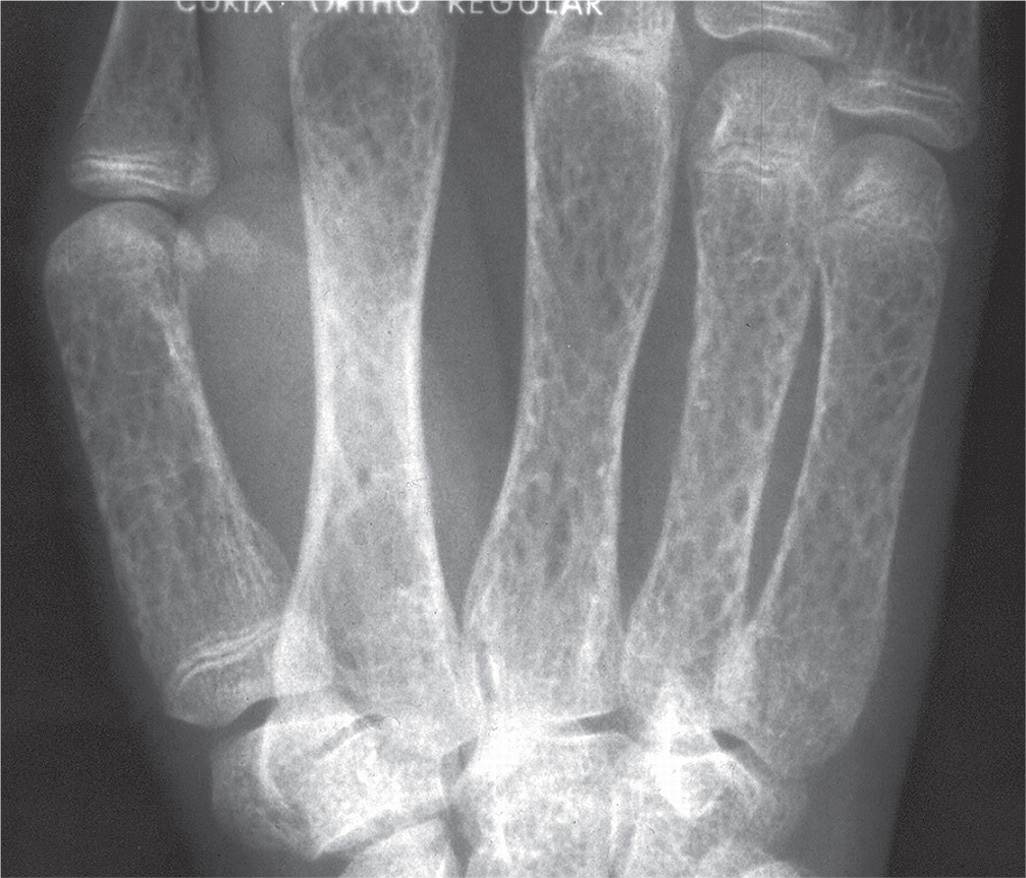
FIGURE 21-14. Thalassemia major.
GENERALIZED SCLEROSIS
Four skeletal dysplasias are the result of abnormal metaphyseal and periosteal ossification caused by either excessive osteoid production or deficient osteolysis. The primary radiographic feature is increased bone density. They include osteopetrosis, melorheostosis, osteopoikilosis, and osteopathia striata. Other disorders demonstrating diffuse sclerosis include pyknodysostosis, fluorosis, and hypervitaminosis D.
Osteopetrosis
Osteopetrosis, which literally means “stone bone”, has also been referred to as marble bone disease. It was originally known as Albers-Schönberg disease; however, since that time, at least 20 different types of osteopetrosis have been described, all of them showing widespread osteosclerosis and increase in overall skeletal mass.7,61 It has been determined that the previously reported Albers-Schönberg disease is the autosomal dominant inheritance type.62
Osteopetrosis is believed to be a consequence of defective osteoclast function coupled with a defect in bone resorption, resulting in increased bone density and reduction in bone marrow spaces.61 There is an inability to differentiate between the cortex and the medullary cavity in tubular bones.63 As excessive bone is formed, the involved bone becomes denser but more brittle.
Traditionally, osteopetrosis has been divided into two forms: the “malignant” or “congenita” autosomal recessive osteopetrosis (ARO), with severe early symptoms, and the “benign” or “tarda” autosomal dominant osteopetrosis (ADO), with mild symptoms.7,62 ARO occurs in infancy and results in death by 10 years of age. The ADO form is of two different types (ADO type I and type II), and patients have normal life expectancy. About 40% of patients with ADO are symptom free regardless of type. Bone pain is common to both types.63 Osteopetrosis is diagnosed almost exclusively on clinical and radiologic evidence.
The ARO type demonstrates frequent pathologic fractures. Radiographically, the ends of long bones are sclerotic and the metaphyseal regions expand, sometimes appearing in the shape of an “Erlenmeyer flask.” There is variable appearance of endobone (“bone within bone”) and transverse bands of sclerosis.62
ADO type I is a relatively benign form. The fracture rate is low because of the increased bone strength compared to normal.63 Extremity involvement is not common but, when present, demonstrates diffuse sclerosis and cortical thickening of tubular bones,
Most commonly encountered is ADO type II, which may be asymtomatic in 20% to 40% of patients; however, these patients are prone to fracture in the absence of significant trauma due to decreased resistance to torsional forces.61 The bones can have increased resistance to compression, causing brittleness during surgery and increased rate of surgical complications, including nonunion, infections, prosthesis loosening, and intraoperative fractures.64 Lower limb deformities (coxa vara; genu valgum or genu varum; clubfoot) have been associated. Pathologically, bone appears disorganized, with rare and short collagen fibers.65 Serum acid phosphatase level is greatly increased in ADO type II, in contrast to ADO type I where it is normal.63
Radiographically, ADO type II osteopetrosis presents with diffuse bone sclerosis (Figure 21-15). Its characteristic feature has been described as endobone or the “bone within a bone” appearance (i.e., the shadow of a smaller template of the bone is seen inside of a tubular, flat, or irregular bone).62 Sclerotic transverse bands can occcur at the femoral and tibial metaphyses, in children and adults alike.61,62 Coxa vara may be found in up to 17% of cases, according to Stoker.62
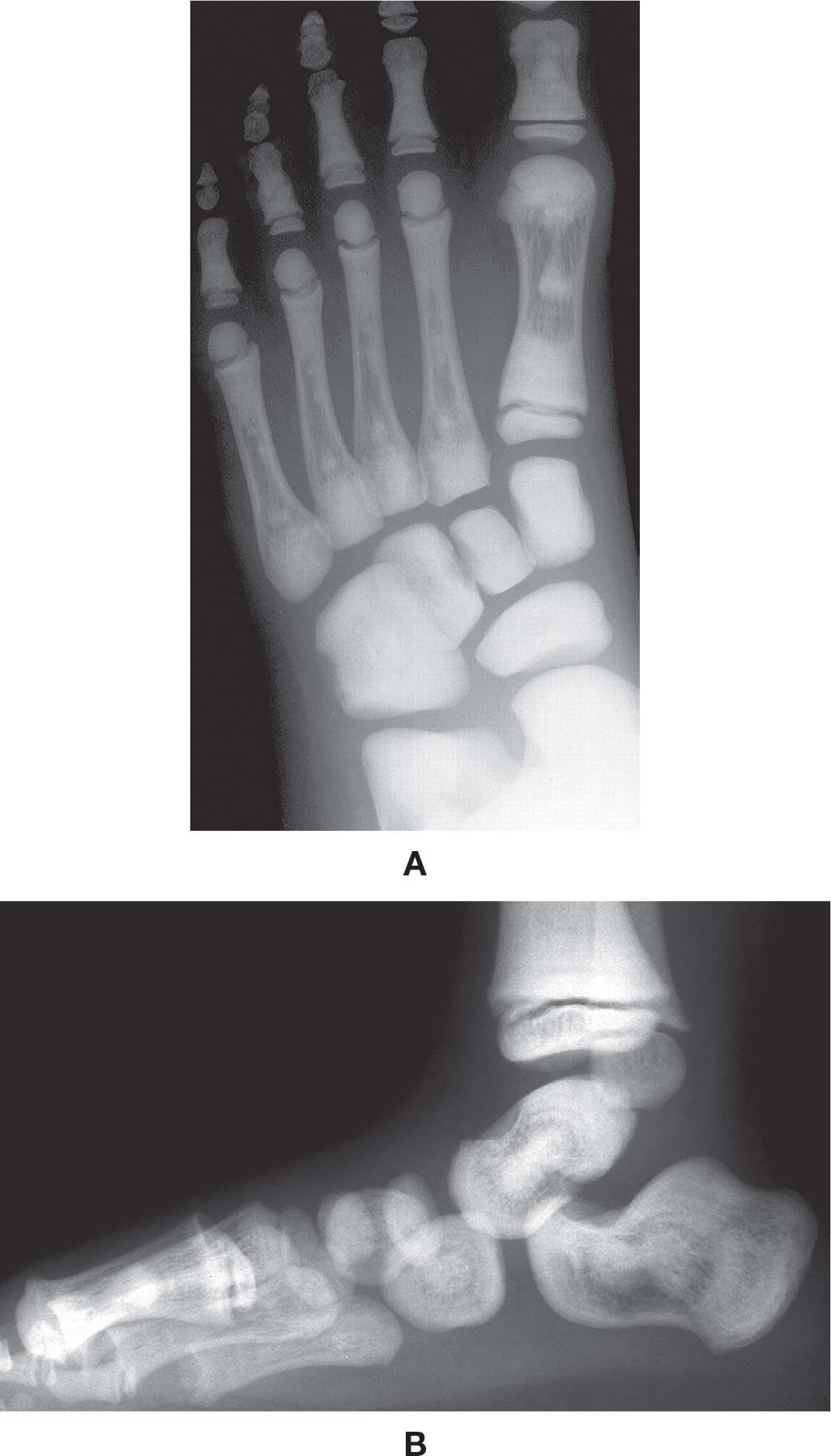
FIGURE 21-15. Osteopetrosis. A: DP view, 7-year-old. B: Lateral view, same patient at 3 years of age. Classic endobone (“bone within bone”) appearance.
Melorheostosis
Melorheostosis (Leri disease) is a rare dysplasia, characterized by a “flowing” hyperostosis along the cortex of tubular bones”.66 It may not be an inherited disorder, according to the literature.3 Melorheostosis is usually asymptomatic but can cause limb stiffness and pain; it has also been associated with skin changes (sclerodermatous skin lesions), hemangiomas, circulation problems (lymphedema), joint contractures, muscle atrophy, and functional limitation.50,66
Melorheostosis is a chronic progressive disorder, and the severity of symptoms often depend on the age of onset; it occasionally results in substantial disability that may lead to amputation.67 It is typically limited to the extremities, affecting a single bone (monostotic), multiple bones (polyostotic), or a single limb (monomelic). The distribution in the affected bones is thought to be due to the predilection of melorheostosis to occur in sclerotomes (skeletal regions innervated by a single spinal sensory nerve).68
The radiographic presentation of melorheostosis is typically found incidentally and consists of hyperostosis typically along one side of a bone’s periphery, extending along its entire length in many cases (Figure 21-16). This picture simulates wax flowing or dripping down the side of a candle and has a wavy, sclerotic bony contour; when present along the cortical endosteal surface, it may partially or fully obliterate the medullary canal.69 Besides the classical flowing hyperostosis appearance, another four radiographic patterns have been described in the literature for this disease: osteoma-like; myositis ossificans-like; osteopathia striata-like; and mixed, with possible overlap syndromes.67
According to Bansal,67 the radiographic findings may reflect a developmental error primarily in intramembranous bone formation, leading to an irregular thickening of cortical bone (cortical hyperostosis) that extends up to the articular surface without passing it. Pathologic findings suggest both overproduction of bone matrix and increased angiogenesis.
Although other imaging techniques rarely are needed for the diagnosis of melorheostosis, similar findings were noted on CT scans. MRI shows decreased signal intensity with all pulse sequences in the affected bones, while radionuclide bone scanning shows moderate and asymmetric increased uptake.67
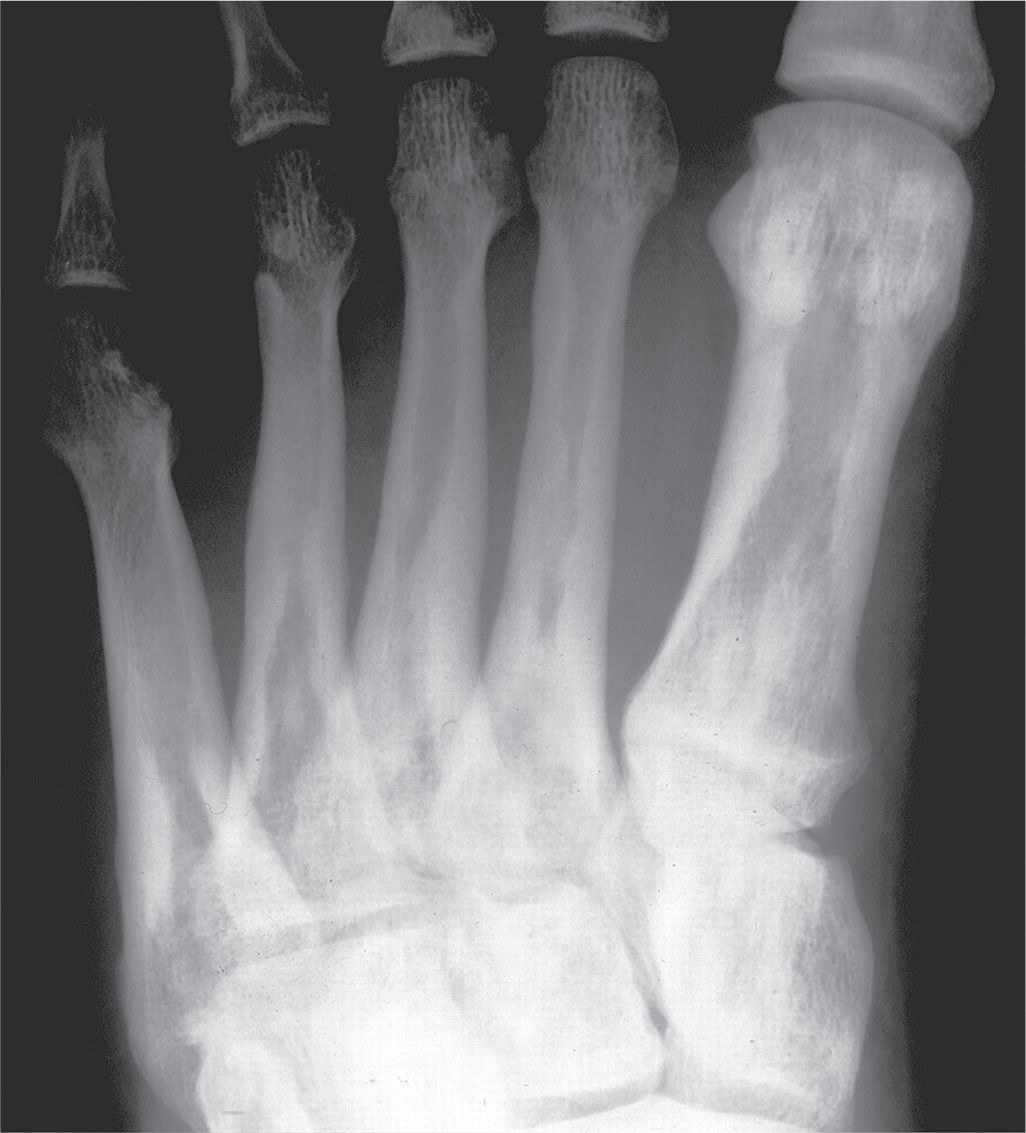
FIGURE 21-16. Melorheostosis. The wavy, thickened endosteal cortical surface simulates wax flowing down a candle. This patient also has osteopoikilosis, as demonstrated by the circular increased densities in periarticular regions.
Osteopoikilosis
Osteopoikilosis is an inherited, autosomal dominant skeletal dysplasia that is not associated with symptomatology. It can occur either as an isolated disorder, or in association with other abnormalities of skin and bone.66 When associated with disseminated connective tissue nevi, it is called Bushke Ollendroff syndrome. Rarely, it may associate with melorheostosis (Figure 21-16).66 Its etiology is not entirely known, but Helleman has found that it is probably caused by a loss-of-function mutation in a gene.66 In osteopoikilosis unequal distribution of numerous small, well-defined and homogeneous circular foci of increased density (hyperostotic areas) are seen radiographically in different parts of the skeleton, mimicking multiple bone islands (Figure 21-17).66,70 The distribution of these lesions is periarticular and symmetric, found at the ends of long bones.3 They may also be found in tarsal bones.71 These lesions, usually detected incidentally, represent foci of old remodeled bone with lamellar structure, either connected to adjacent trabeculae of spongy bone or attached to the subchondral cortex.66
Osteopathia Striata
Osteopathia striata is most likely inherited, possibly due to mesodermal mosaic dysfunction; it may be seen in childhood, adolescence, and adulthood, and is typically asymptomatic.50,72 Radiographically, linear and regular, fine bands of increased and decreased density extend from the metaphysis to the diaphysis, running parallel to the shaft of long bones (Figure 21-18); it is of no clinical or medical significance.73 It is not clear if the striations reflect increased or decreased bone density, and it is also unclear if these individuals are at increased risk for generalized osteoporosis and spontaneous fractures.73
Pyknodysostosis
Pyknodysostosis is an autosomal recessive disorder, thought to be caused by a chromosome mutation that will lead to diffuse sclerosis of bone.3,23,74 It is associated with recurrent fractures and dwarfism and, as a result, is diagnosed at an early age. Radiographically there is generalized increased bone density (osteosclerosis) and narrowing of the medullary canals. Hypoplasia or acro-osteolysis of the distal phalanges are characteristic features, as is increased bone fragility.74
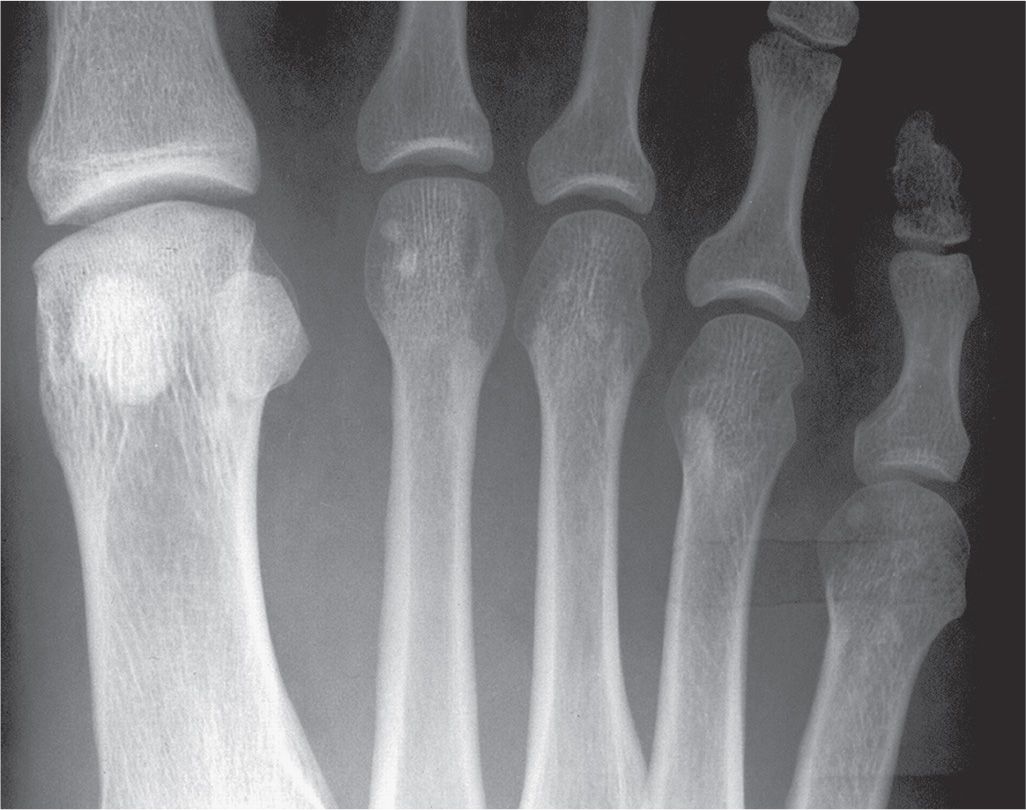
FIGURE 21-17. Osteopoikilosis. Multiple islands of cortical bone density are identified in the lesser metatarsal heads.
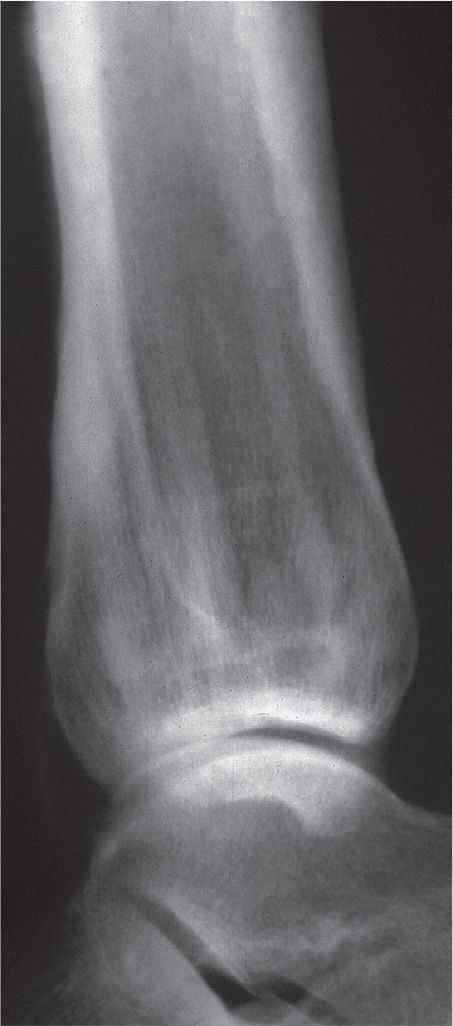
FIGURE 21-18. Osteopathia striata. Lateral view, ankle. Mixed linear bands of increased and decreased density run through the metadiaphyseal region of the distal tibia, parallel to the bone’s long axis.
Fluorosis
Fluorosis results from chronic fluorine intoxication. Nearly all of the fluorine that is not excreted by the kidneys (50%) is retained in calcified tissue. Clinical manifestations of prolonged exposure may include joint pain, limited range of motion, and palpable thickening of the tibia.3
Radiographic findings of skeletal fluorosis in the appendicular skeleton may include osteopenia (which is prone to fractures), ligament calcification, enthesopathy, and thickening of the periosteal surfaces of bones.3 Classic skeletal fluorosis is a far advanced manifestation of the disease, and may present abnormal fracturing of bone with a generalized distinctive increase in bone density.75
Hypervitaminosis D
Overdose of vitamin D can be acute or chronic. It occurs with prolonged intake of more than 50,000 IU/day. The increased concentrations of vitamin D metabolite reaches the vitamin D receptor in the nucleus of target cells, causing exaggerated gene expression.76 Hypervitaminosis D causes hypercalcemia, hyperphosphatemia, and normal or low PTH level. Clinically it may lead to painful periarticular calcinosis.76
Hypervitaminosis D also causes a severe disruption of enchondral bone formation, with excessive calcification of the proliferating cartilage cells, causing widening of the zone of provisional calcification.7 Excess vitamin D ingestion by infants is related to chronic idiopathic hypercalcemia. Radigraphically this causes a generalized increase in bone density. Increased calcification can be seen in the soft tissues, including blood vessels and around joints.
GENERALIZED PERIOSTITIS
Generalized periostitis is associated with venous stasis, hypertrophic osteoarthropathy, hypervitaminosis A, and thyroid acropachy.3,77 Radiographically, varying degrees of periosteal new bone formation is possible, but typically it is thick and undulating (irregular). It also may be seen in the small tubular bones of the foot with tuberous sclerosis.
Venous Stasis (Insufficiency)
10% to 35% of United States adults have some form of chronic venous insufficiency, which seems to be autosomal dominant with variable penetrance mode of genetic transmission; however, the chromosome responsible for the disease is currently unknown. It has a decreased incidence in males, and causes chronic ambulatory venous hypertension with varicose vein formation (90% risk when both parents are affected, 25%–62% risk if only one parent is affected).78
The chronic inflammation noted with venous insufficiency has been associated with periosteal new bone formation, which increases with the severity of venous insufficiency. Periostitis may be seen in 10% to 60% of patients with severe venous stasis.3 This is often an incidental finding and may be caused by periosteal hyperemia, though the underlying mechanism remains unclear.79,80 Soft tissue calcification is a common associated finding.3 The tibia is a frequent location (Figure 21-19).
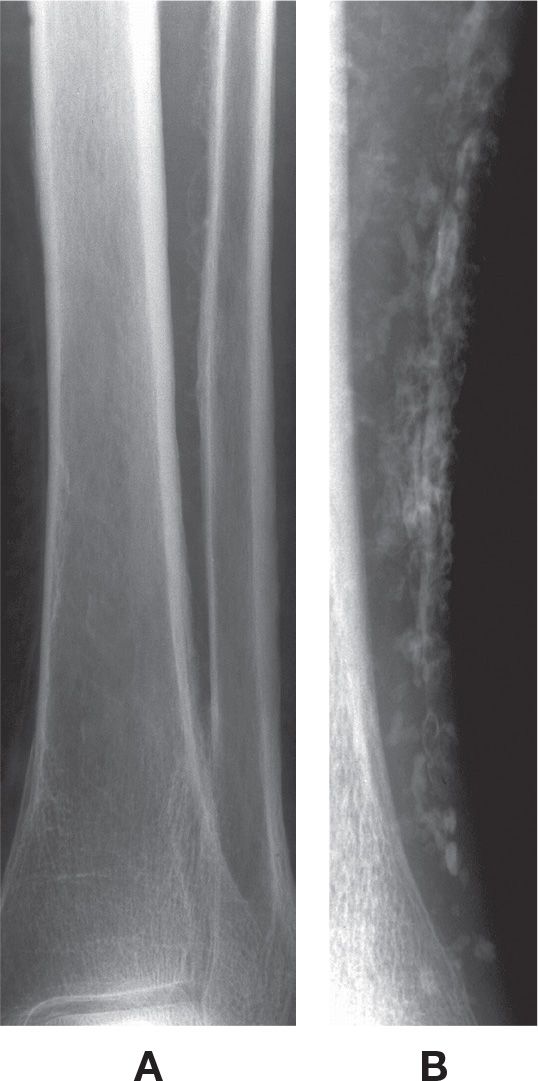
FIGURE 21-19. Generalized periostitis: venous stasis. A: Irregular, wavy periostitis is identified along the lateral margin of the distal tibia and medial margin of the fibular diaphysis. B: This patient, with chronic venous insufficiency (AP view), demonstrates solid periostitis along the distal tibial diaphysis with diffuse soft tissue calcification (phleboliths).
Hypertrophic Osteoarthropathy
Hypertrophic osteoarthropathy refers to a clinical syndrome that consists of digital clubbing, extremity enlargement and swollen joints, without signs of acute inflammation.3,81,82 The disease takes its name from the enlargement of bones seen in later stages.81 Radiography is often used to diagnosis hypertrophic osteoarthropathy, which is divided into two forms, primary and secondary.
Primary (hereditary or idiopathic) hypertrophic osteoarthropathy (also known as pachydermoperiostosis) is hereditary with autosomal dominance of variable penetrance and accounts for 3% to 5% of cases of hypertrophic osteoarthropathy. It has an insidious onset and a predilection for black adolescent males (M:F ratio of 7:1).82 Although the diagnosis is based on its classic clinical and radiologic features, it is often missed due to its variability in presentation. Reports include increased density of tarsal, metatarsal, and phalangeal bones, with joint space narrowing of interphalangeal joints; periarticular osteopenia and resorption of the distal phalanges, associated with soft tissue swelling of the distal toes and contractures of the toes; and enlargement of the sesamoid bones, joint deformity, and calcaneal collapse.82 The predominant finding, periostitis, causes a thickened and sometimes shaggy appearing cortex that extends to the epiphysis.3
Secondary hypertrophic osteoarthropathy results from cardiopulmonary disease, hepatic disease, gastrointestinal disease, and certain malignancies.82 It is sometimes called pulmonary hypertrophic osteoarthropathy due to the association of periostitis with disorders such as bronchogenic carcinoma (5% of cases).3,81 Unlike pachydermoperiostosis, secondary hypertrophic osteoarthropathy demonstrates periostitis that is distinct from the underlying cortex; it is more linear than it is shaggy or irregular, which is characteristic of the primary form. It is also associated with digital clubbing and joint abnormalities. It occurs at the ends of the tibia and fibula and along the diaphysis of metatarsals and phalanges. Patients may experience a dull ache with accompanying synovitis.
Hypervitaminosis A
Increased levels of vitamin A for any reason (treatment or consumption) will stimulate periosteal new bone formation, which will cause cortical thickening; it has a predilection for the metaphysis.83,84 Radiographic features are limited to children. Soft tissue nodules will be found adjacent to cortical abnormalities.3
Thyroid Acropachy
Thyroid acropachy is a rare manifestation of an autoimmune associated thyroid disease with polygenic predisposition.2 It has an incidence estimated to be approximately 1% of patients that have thyrotoxicosis (Graves disease), but has also been described in patients with Hashimoto thyroiditis and in euthyroid patients.3,85 Graves disease is the second most common autoimmune thyroid disease, following only Hashimoto thyroiditis, and is the most common cause of hyperthyroidism in noniodine deficient areas.86 Tobacco use is found to be commonly associated with acropachy (75% in man and 81% in women).86
The clinical manifestations include clubbing and skin tightness of the digits, soft tissue swelling, and pain in distal small joints.85 In most cases of acropachy, digital clubbing is asymptomatic. In more advanced cases, fusiform swelling of the digits is noted. About 20% of patients with dermopathy have acropachy. The presence of acropachy and dermopathy are predictors of severity of the autoimmune process.86
The most distinguishing radiographic feature of acropachy is a solid periosteal reaction that may appear thick or thin. The periostitis can present as feathery, lacy, frothy, or shaggy; it may be spiculated and perpendicular to the shaft. It tends to be bilateral and involves the tubular bones of the foot, sparing the ends of bones.85 The reaction often is mid-diaphyseal and symmetrical. The bones most commonly involved are the short tubular bones of the foot, often involving the proximal and middle phalanges of the same digit and the first metatarsal. Less commonly, thyroid acropachy can involve the long bones of the leg.
The periostitis is helpful in confirming thyroid acropachy, but it is not necessary for diagnosis.85 Radiographic evidence of periostitis can be confirmed with bone scintigraphy, which shows increased uptake along the periosteum. MRI features of dermopathy and acropachy have been described in the literature, but MRI is not needed for diagnosis.86
Tuberous Sclerosis
Tuberous sclerosis is an autosomal dominant disease, remarkable for its genetic heterogeneity, high spontaneous mutation rate, and extremely variable and age-dependent clinical phenotype. The classic triad of mental retardation, epilepsy, and hamartomas presents clinically.87 Radiographically, it produces a wavy periostitis of metatarsals and phalanges. Other skeletal findings include patchy increased densities that are island-like as well as lucent areas in the diaphyses.3
SOFT TISSUE MANIFESTATIONS OF SYSTEMIC DISORDERS
The primary radiographic soft tissue findings associated with systemic disorders are calcification and ossification. Calcification appears as irregular punctate, circular, linear, or plaque-like radiodense areas with no cortical or trabecular structure.3 Ossification, in contrast, demonstrates a trabecular pattern and a thin, cortex-like periphery. However, if the lesion is small, the distinction may be impossible to determine visually.
Calcification
Conditions that lead to soft tissue calcification have been categorized as metastatic calcification, generalized calcinosis, and dystrophic calcification.3 Metastatic calcification (Figure 21-20) occurs in normal tissue and results from a disturbance in calcium or phosphorus metabolism. Examples include hyperparathyroidism, hypoparathyroidism, renal osteodystrophy, hypervitaminosis D, and sarcoidosis.
Generalized calcinosis (Figure 21-21) presents as calcium deposition in the skin and subcutaneous tissue in the presence of normal calcium metabolism. There are three types of calcinosis: calcinosis circumscripta, calcinosis universalis, and tumoral calcinosis.
Calcinosis circumscripta is associated with collagen vascular disease. Dermatomyositis demonstrates fine, often reticular-like calcification. The calcinosis seen in scleroderma has a diffuse linear, streaky pattern. It can involve the Achilles tendon and the forefoot region.
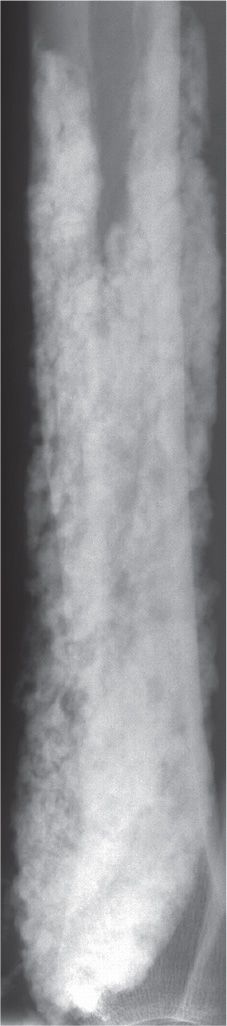
FIGURE 21-20. Metastatic calcification: secondary hyperparathyroidism. The spectrum of radiographic abnormality may be seen in these patients. This example demonstrates significant soft tissue calcification in the distal leg.
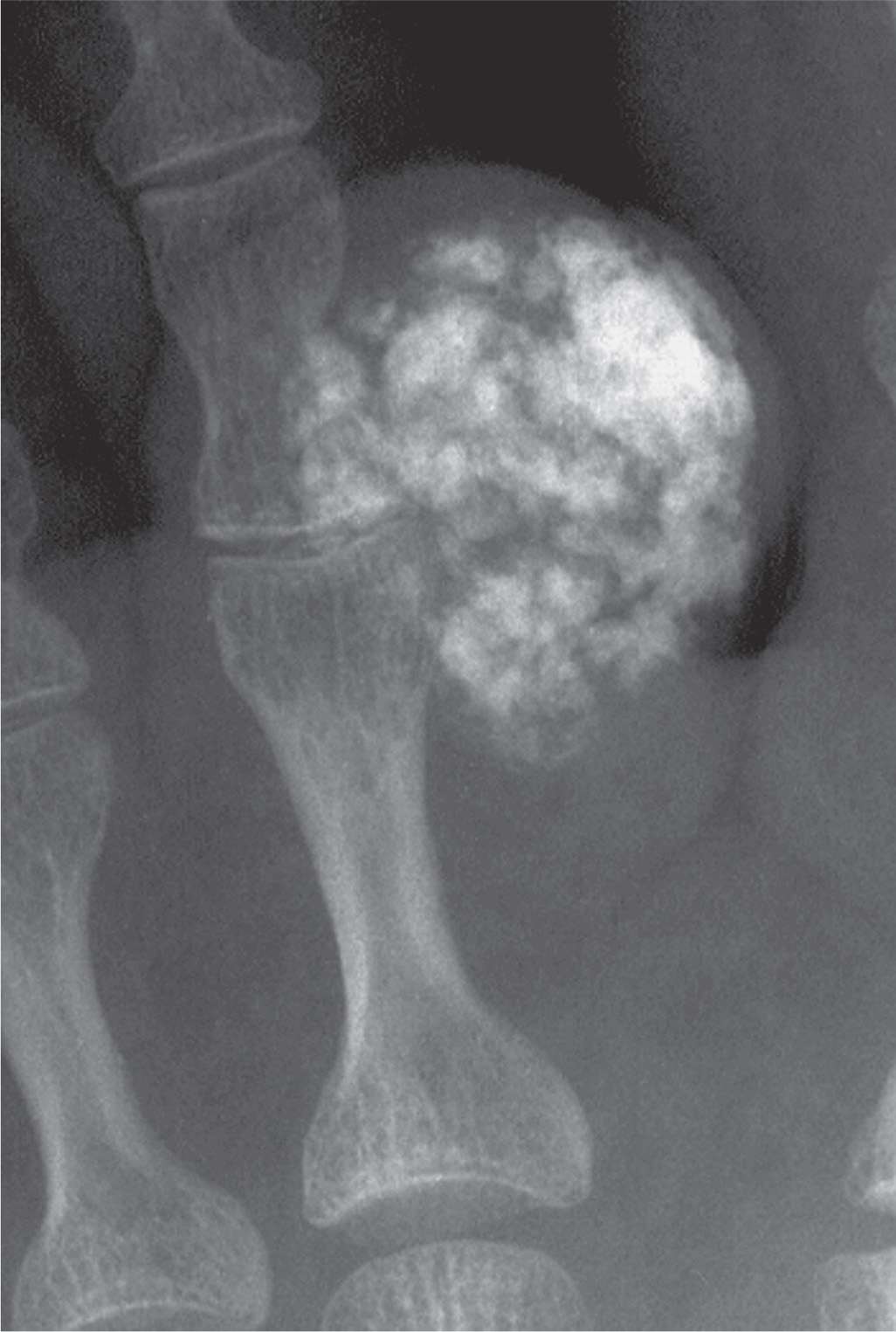
FIGURE 21-21. Generalized calcinosis: tumoral calcinosis. A large periarticular mass demonstrates calcification adjacent to the proximal interphalangeal joint of a toe.
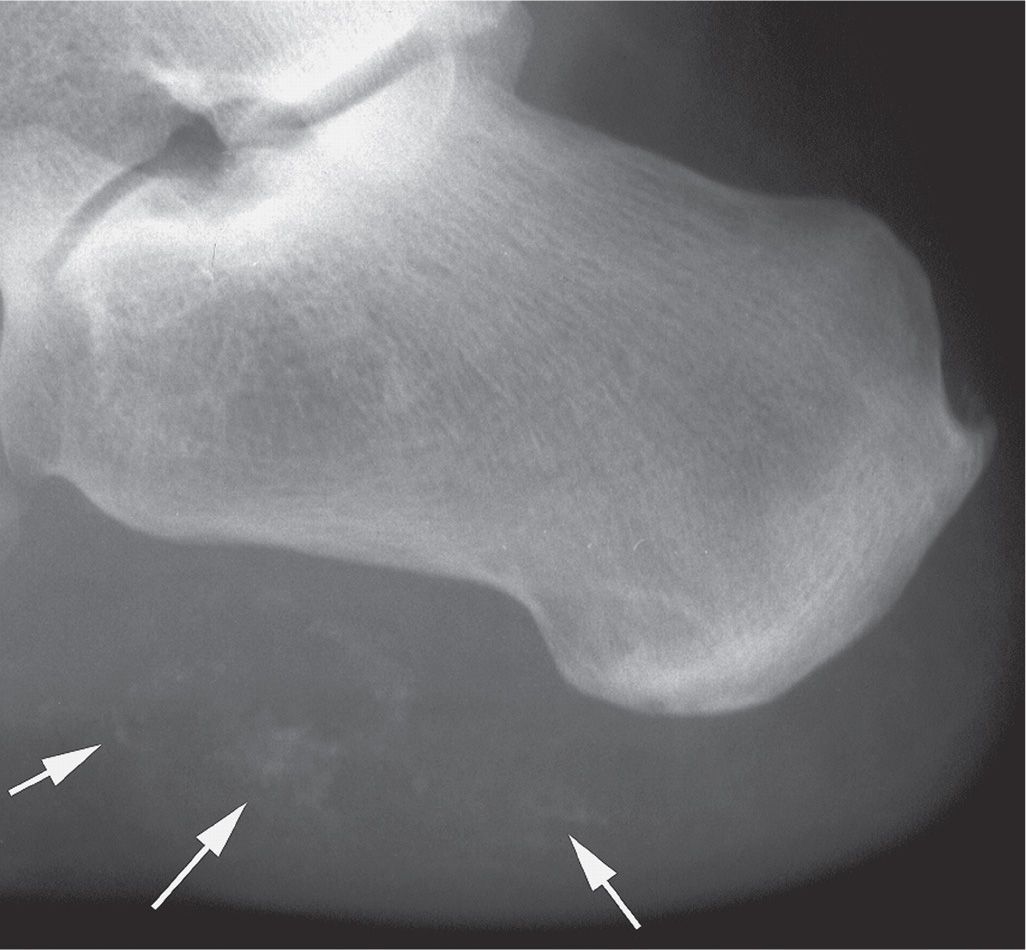
FIGURE 21-22. Dystrophic calcification: trauma. This patient related a recent injury to the heel. Multiple areas of ill-defined soft tissue calcification are identified (arrows).
Calcinosis interstitialis universalis effect younger ages and demonstrates a longitudinal streaky appearance.
Tumoral calcinosis is a relatively rare disorder most commonly seen in people of African descent.88 The etiology is uncertain. The histologic appearance is characterized by densely loculated masses of calcific debris and fluid enclosed by fibrous tissue (Figure 21-21).89 Tumoral calcinosis is rare in the foot but has been reported adjacent to a sesamoid and a first metatarsal head medial eminence, at the tip of digits, and posterior to the ankle joint.89,90 The radiographic hallmark is a large, periarticular mass, usually along the joint’s extensor surface, that is calcific and multinodular.88
In dystrophic calcification (Figure 21-22), calcium is deposited in damaged or devitalized tissue in the absence of a generalized metabolic derangement; this is the most common type of calcification seen in the foot and ankle. Examples include neoplasm, inflammation, and trauma; Table 21-4 provides a useful mnemonic for its differential diagnosis. Soft tissue calcification has been associated with local corticosteroid injection of the heel and intra-articular corticosteroid injection of the small joints of the hand.91,92 Extensive involvement of the foot suggests sarcoma.93
| Differential Diagnosis for Dystrophic Calcification: VINDAT |
| Example |
Vascular | Phlebolith |
Infection | — |
Neoplasm | Osteosarcoma |
Drugs | Vitamin D |
Autoimmune | Dermatomyositis, scleroderma |
Trauma | — |
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


