20 TRAUMATIC BRAIN INJURIES
Brain injury is the leading cause of all trauma-related deaths. It constitutes the primary cause of trauma-related death and long-term disability among young Americans.1,2 Survivors of the initial brain insult remain at risk for various multisystem complications that can exacerbate the initial brain injury and increase mortality and morbidity. The prevention, recognition, and immediate treatment of these complications in the prehospital, resuscitation, critical care, intermediate care, and rehabilitation phases of care are of utmost importance. Recovery is often a lifelong process beset with physical, mental, emotional, and social obstacles. Brain injury and the potential long-term disabilities it creates also have a tremendous impact on the family and cost society billions of dollars each year. Because of its high incidence, mortality and morbidity, economic cost, and demand for medical resources, brain injury is a major public health problem in the United States.
EPIDEMIOLOGY
The precise number of TBI cases is not known; however, it is estimated that about 1.4 million Americans sustain TBI each year.1 In 1996 the Centers for Disease Control and Prevention (CDC) was charged by Congress with developing a uniform reporting system to track the incidence, severity, causes, and outcomes of TBI (Public Law 104-166).3 In response the CDC established guidelines for surveillance of central nervous system injury and published “Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths”1 and “Incidence Rates of Hospitalization Related to Traumatic Brain Injury—12 States, 2002.”4 These reports are limited in that the former only provides data about patients seen in a tertiary system and the later reports data from only 12 states.
“Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths” reports that from 1995 to 2001, there were on average 1,111,000 emergency department (ED) visits for TBI, 235,000 hospitalizations, and 50,000 deaths per year. Falls are the leading cause of TBI, followed by motor vehicle crashes.1 Of all reported TBIs, approximately 398,000 are related to falls, 280,000 to motor vehicle traffic incidents and 156,000 to assaults.2 Each year, an estimated 80,000 to 90,000 Americans sustain permanent disability from their TBI.2 At least 5.3 million Americans are currently living with disabilities resulting from TBI.1
The Traumatic Brain Injury Model Systems National Data Center, a division of the National Institute on Disability and Rehabilitation Research—United States Department of Education, established the TBI National Database.5 Patient information for this database was contributed by 21 federally funded TBI Model Systems of Care throughout the United States. As of September 2005, the database contained information on 5756 persons age 16 and older with primarily moderate to severe TBI. More than 62% of the patients had a Glasgow Coma Scale (GCS) score ranging from 3 to 8, were intubated, or in chemical coma. According to this database the mean age of patients with TBI is 38.6; 69% are non-Hispanic Caucasian, 20% African-American, 7% Hispanic, and 2.7% Asian/Pacific Islander. Of cases admitted to the Model Systems facilities, motor vehicle crashes were the most common cause of TBI (46.9%) followed by falls (19.5%). Since 1997, 7.7% of the database sample suffered TBI from blunt assaults. More than half (63%) of the individuals were employed at the time of their injury, and by post-injury year 10, only 29% were employed. After sustaining TBI, mean length of stay for acute hospitalization was 20.0 days and 26.3 days for rehabilitation. It is important to remember that these statistics only reflect data based on a sample of patients with TBI. The true numbers of those who have sustained a TBI are not known.
The economic costs of TBI are staggering. Direct costs include initial and follow-up diagnoses, treatment, and rehabilitation. Indirect costs include societal losses secondary to restricted or lost productivity. In the United States, the estimated cost of TBI totaled an estimated $60 billion for the year 2000.6 However the physical and psychosocial suffering endured by brain-injured persons and their families is incalculable.
CORRELATIVE NEUROANATOMY AND PHYSIOLOGY
The brain, which is part of the central nervous system, provides most of the control functions for the entire body. The brain receives and interprets sensory input and then integrates this information to determine and carry out the response to be made by the body. In general the brain maintains the quality and uniqueness of human life and behavior. The major divisions of the brain are the cerebrum, brainstem, and cerebellum (Figure 20-1), which are housed within multiple protective coverings.
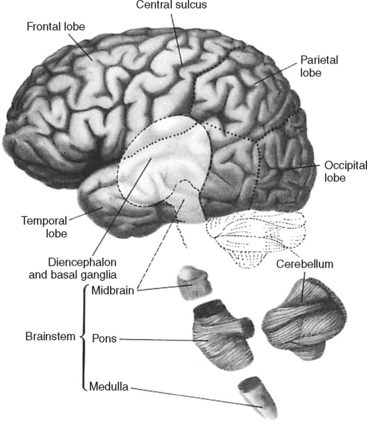
FIGURE 20-1 Major parts of the central nervous system.
(From Kandel E, Schwartz J: Principles of Neural Science, ed 3, New York, 1991, Elsevier/North-Holland.)
COVERINGS OF THE BRAIN
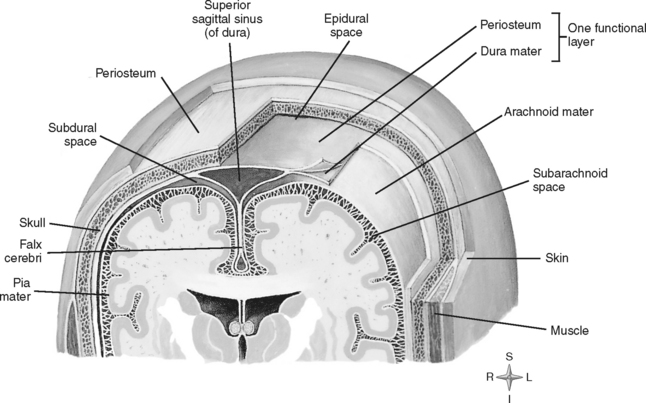
Numerous protective coverings surround the brain. Outermost on top of the skull is the scalp, composed of multiple layers: skin, subcutaneous fascia, galea aponeurotica, and periosteum. The rigid, nondistendible cranium that encases the brain is made up of the sphenoid, ethmoid, frontal, and occipital bones, two parietal bones, and two temporal bones (Figure 20-2). Beneath the skull are three layers of connective tissue (the meninges) that surround the brain and the spinal cord. The meningeal layers include the outermost thick, fibrous dura mater; the fine, elastic arachnoid mater; and the innermost vascular membrane, the pia mater, which adheres to the brain’s surface (Figure 20-3).
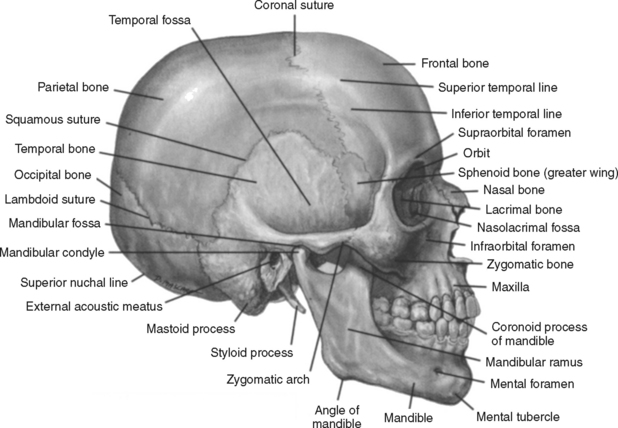
FIGURE 20-2 Right side view of the skull.
(From Lindsay DT: Functional Human Anatomy, St. Louis, 1996, Mosby, p. 147.)
CEREBRUM
Cerebral Hemispheres
The two cerebral hemispheres are divided into four pairs of lobes: the frontal, temporal, parietal, and occipital (Figure 20-4). Knowledge of the primary functions of the lobes (Table 20-1) and lesion location can alert the nurse to the most likely assessment findings. For example, it can be anticipated that a focal lesion in the prefrontal region may result in altered behavior, whereas a lesion in the posterior frontal region of the dominant hemisphere may result in language dysfunction. Nursing assessment and the patient plan of care should anticipate and focus on the actual or potential problems associated with lesions in specific anatomic locations.
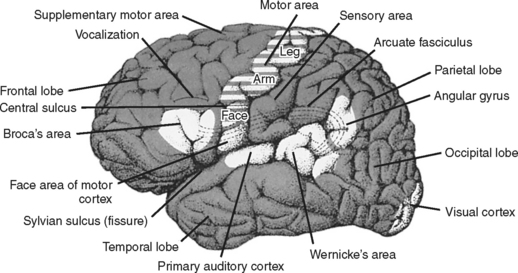
FIGURE 20-4 Lobes of the brain.
(From Kandel E, Schwartz J: Principles in Neural Science, ed 3, New York, 1991, Elsevier/North-Holland.)
TABLE 20-1 Functional Localization in the Cerebral Cortex
| Lobe | Functions |
|---|---|
| Frontal | |
| Temporal | |
| Parietal | |
| Occipital |
* Cerebral dominance: In right-handed and most left-handed people, the left cerebral hemisphere is dominant for language, mathematical, and analytic functions. The opposite nondominant hemisphere is thought to be concerned with nonverbal, geometric, spatial, visual, and musical functions.
Used with permission from McQuillan KA: The neurologic system. In Alspach GA, editor: Core Curriculum for Critical Care Nursing, ed 6, St. Louis, 2006, Saunders Elsevier, p. 384.
The basal ganglia and limbic system, which provide a number of important functions, are located deep within the cerebral hemispheres. The basal nuclei are masses of gray matter, (i.e., caudate, putamen, claustrum, amygdaloid, globus pallidus, and functionally, the substantia nigra and subthalamic nuclei), which control and regulate coordination of motor integration, voluntary movement, motion initiation, postural reflexes, and muscle tone. The basal ganglia serve as the principle center of the extrapyramidal motor system. The limbic system is composed of the limbic lobe and several other structures with which it is functionally and anatomically related, including the hippocampus, amygdala, fornix, olfactory tract, thalamus, and hypothalamus. This system is responsible for some aspects of learning and memory, instinctual drives (e.g., hunger, mating), and emotional behavioral responses and associated endocrine, visceral, and somatic reactions.
BRAINSTEM
The brainstem is a midline structure situated beneath the diencephalon. The brainstem has three sections: the midbrain is the uppermost structure, the pons lies in the middle, and the medulla is the lowest segment (see Figure 20-1). The descending medulla is contiguous with the spinal cord, which begins at the level of the foramen magnum.
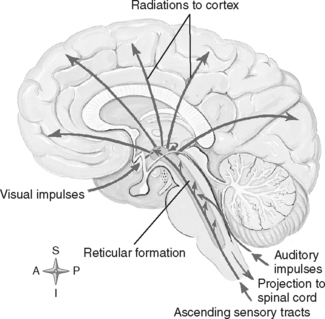
The brainstem performs a variety of vital functions. It is the center of the brain that controls basic and reflexive activities, such as visual and auditory motor reflexes, wakefulness, sneezing, coughing, breathing, vasomotor activity, and heart rate regulation. It is the pathway used by sensory fibers as they ascend from the cord to the cortex. Motor tract fibers on their descent from their cerebral or brainstem origin also traverse the brainstem, and many cross over (decussate) in the medulla before exiting down the spinal cord. Many cerebellar tracts also pass through the brainstem, which allows formation of extensive connections between the cerebrum and cerebellum in this region. The reticular activating system responsible for consciousness originates in the brainstem as a dense bundle of fibers and ascends, coursing out over the cerebral cortices (Figure 20-5). The brainstem also controls many of the special senses, motor functions, and reflexes that are mediated by the cranial nerves, which arise and exit from this structure.
CEREBROSPINAL FLUID
The CSF pathway (Figure 20-6) is a free-flowing system whereby CSF passes from the lateral ventricles (hollow cavities within each cerebral hemisphere) through the foramina of Monro (intraventricular foramina) into the third ventricle. From there it flows into the fourth ventricle via a small, narrow opening known as the aqueduct of Sylvius. CSF exits the fourth ventricle through the foramina of Magendie and Luschka to enter the cisterna magnum and the subarachnoid space (SAS). CSF within the subarachnoid space flows upward over the convexity of the brain and downward around the spinal cord.
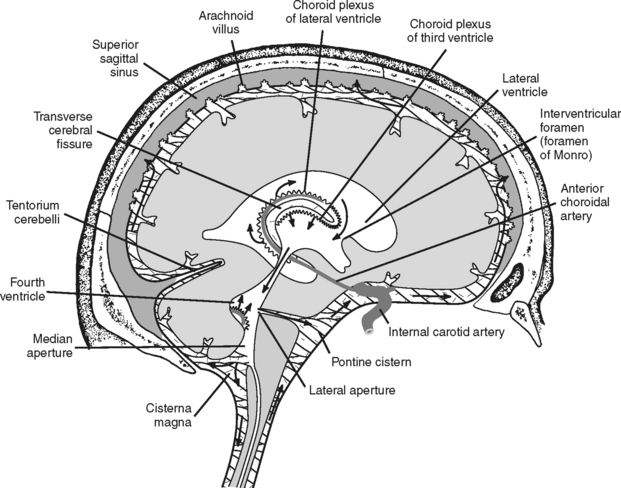
FIGURE 20-6 Path of CSF circulation.
(From Nolte J: The Human Brain: An Introduction to Its Functional Anatomy, ed 4, St. Louis, 1999, Mosby, p. 107.)
CELLS OF THE BRAIN
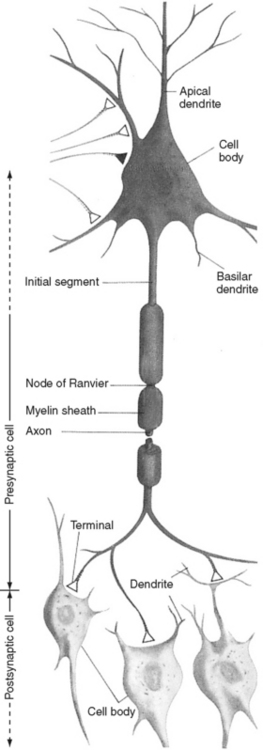
The neuron is the functional unit of the nervous system that transmits nerve impulses. This type of cell consists of a cell body, axon, presynaptic terminals of the axon, and dendrites (Figure 20-7). Each of these cell components has a distinctive function. The cell body synthesizes and packages the products of neuronal metabolism and transports them to other regions of the cell. It exchanges nutrients, ions, and other metabolically active substances with the extracellular environment through the cell membrane. This lipoprotein membrane is crucial for depolarization of the neuron through such mechanisms as the energy-dependent sodium-potassium pump, ion-selective channels, and voltagesensitive channels. The myelin-covered axon, a tubular extension of the cell body, is responsible for rapid nerve conduction away from the cell body. The presynaptic terminals are specialized endings at the distal portion of the axon that transmit information by chemical or electrical means to other neurons or effector cells. The specialized contact zone between the transmitting (presynaptic cell) and the receiving (postsynaptic cell) neuron is the synapse. The dendrites, unmyelinated fibers that branch extensively as they extend from the cell body, receive incoming impulses and transmit them to the cell body. The physiologic and anatomic properties of the nerve cell account for its unique ability to communicate with other cells. In general the neuron is responsible for processing, analyzing, and acting on all incoming and outgoing information, which culminates in all our behavioral responses. Neurons differ from other cells in both their communicative ability and their lack of mitotic ability. Without mitosis, neurons cannot multiply. With central nervous system maturity, the number of neurons is fixed and no new neurons are added. Injury to neurons is significant because it results in cell loss with no new cell replacement.
CEREBRAL METABOLISM
The average brain weighs 3 pounds and accounts for 2% of an adult’s body weight. Despite its relatively small size, the brain uses about 20% of the total body oxygen consumption.7 The primary energy source for the brain is glucose, which is converted by aerobic or oxidative metabolism into a high-energy phosphate form, adenosine triphosphate (ATP). Most of the energy is used for neuronal metabolic and conductive activities. Oxygen and glucose are in continuous demand, as the brain has minimal storage capacity for either substrate. The brain’s dependency on energy is so great that, without its nutrient supply, neuronal function fails within seconds of energy deprivation.
The need for oxygen and its rate of delivery depend on the degree of metabolic activity coupled with the rate of oxygen consumption. If metabolism is increased, as occurs with seizures, hyperthermia, or abnormal posturing, oxygen requirements increase. When cerebral metabolism is decreased, such as with sedation or hypothermia, oxidative requirements are reduced.
CEREBRAL BLOOD FLOW
Autoregulatory Mechanisms
Pressure, or myogenic autoregulation, is the brain’s ability to maintain a constant rate of blood flow over a wide range of perfusion pressures despite changes in arterial blood pressure and ICP. The cerebral arterioles or resistance vessels have an inherent self-regulatory (autoregulatory) mechanism contained within their muscular walls that allows them to change vessel diameter in response to changes in transmural pressure (Figure 20-8). As a result, an increase in arterial pressure causes vasoconstriction of the vessels, and a decrease in arterial pressure causes vasodilation. This mechanism keeps cerebral blood flow (CBF) essentially constant for a range of CPP from approximately 50 to 150 mm Hg. Intact autoregulation safeguards the brain from ischemia caused by fluctuations in arterial pressure and ICP.
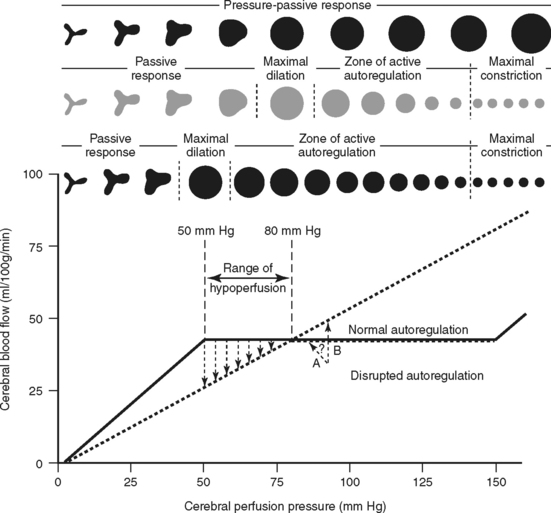
Several metabolic factors also have a marked effect on cerebral blood flow, including partial pressure of arterial oxygen (PaO2), pH, and partial pressure of arterial carbon dioxide (PaCO2). A decrease in PaO2 below 50 mm Hg causes vasodilation, which increases cerebral flow and volume.8 Extracellular reduction in pH (cerebral acidosis) caused by hypercarbia or acid byproducts of anaerobic cellular metabolism induces vascular dilation and increases cerebral flow and volume. Carbon dioxide (CO2) is the most potent vasoactive agent known to cerebral vessels. An increase in CO2 causes vasodilation, increasing cerebral flow and volume. Inversely, a decrease in CO2 constricts the cerebral vessels, decreasing cerebral flow volume. Normocapnic individuals have about a 3% alteration in CBF for every 1 mm Hg change in CO2 between the range of 20 mm Hg and 60 mm Hg.9
Metabolic autoregulation causes the rate of CBF to vary depending on neuronal metabolic activity. Gray matter, which contains the metabolically active cell bodies, normally receives three to six times more flow than white matter. CBF decreases globally in coma and increases regionally with activation or specific activities (e.g., hand movement, mental processing). In coma, both cerebral metabolism and blood flow are normally depressed. Even with a blood flow reduction of as much as 40%, neuronal integrity can still be maintained as long as metabolism is equally reduced.
ETIOLOGY
CAUSES AND RISK FACTORS
Motor vehicle traffic incidents constitute the leading cause of TBIs that result in death or hospitalization, whereas falls are the principle cause of TBI-related ED visits.1 Transportationrelated crashes (involving motor and recreational vehicles, bicycles, and pedestrians) constitute the leading cause of TBI-related deaths and hospitalizations.1 Of all individuals who sustained motor vehicle- or traffic-related TBI, motor vehicle occupants comprised approximately 70% of those requiring hospitalization and more than 50% of those who died.1 Assaults cause 11% of all TBI and 13% of all TBI-related deaths.1 Nine out of 10 people with firearm-related brain injuries die. Nearly two thirds of firearm-related brain injuries are self-inflicted.10
Gender, age, ethnicity, and socioeconomic status can influence the risk of TBI. Males are 1.5 times as likely to sustain a TBI as females, and the death rate from TBIs is 2.8 times higher in males than females.1 Children ages 0 to 4 years, adolescents ages 15 to 19 and adults age 75 and older are more likely than others to sustain a TBI.1 Young children (ages 0 to 4 years) have the highest rate of visits to emergency departments, followed by older adolescents; however, adults older than age 75 have the highest rates of hospitalization and death due to TBI.1 Falls are the most common cause of TBI in persons younger than age 5 and those older than age 75, whereas motor vehicle crashes and assaults are the leading cause of TBI among adolescents and young adults.1 African-Americans reportedly have the highest death rate from TBI, which is primarily due to the high rate of firearm-related deaths in this racial group.1 Lower socioeconomic status may increase the risk of injury because of greater dependence on less safe housing and vehicles, higher likelihood of having a more physically demanding occupation, and increased exposure to personal violence.
MECHANISM OF INJURY
Skull Deformation
Direct impact to the head can distort the skull’s contour and injure the underlying brain tissue (Figure 20-9A). Highvelocity blows to the head are most often associated with deformation injuries. Cranium indentation, fracture, or outward bowing can precipitate contusions or lacerations of the underlying brain tissue and intracranial hemorrhage.
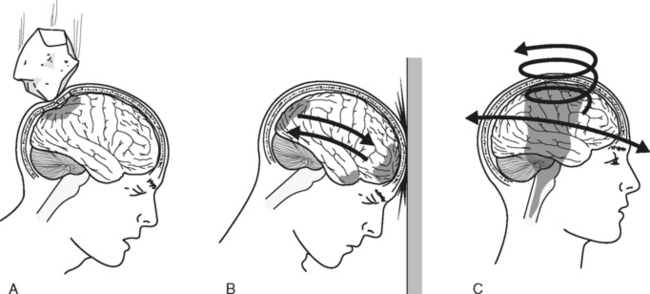
Acceleration-Deceleration Injury
Acceleration-deceleration injuries occur when the head is thrown rapidly forward or backward, resulting in sudden alterations in movement of the skull and brain in a straight, linear path (see Figure 20-9B).11 Acceleration injuries occur when a moving object strikes the head (e.g., a baseball bat, fist, or hammer) and the skull and the brain are set in motion. Deceleration injuries occur when a head that is in motion hits a stationary object such as a wall or windshield, causing the skull to decelerate rapidly. The semisolid brain moves slower than the solid skull; therefore, the brain collides with the cranial surface and the rough bony prominences within the skull, causing brain tissue injury. Injury caused when the brain makes contact at the site of head impact is referred to as coup injury. Brain injury also may occur as the brain is thrown in the direction opposite the impact, causing collision with the contralateral skull surface; this is known as contrecoup injury.
Rotation
Brain injury may also occur when acceleration or deceleration of the brain follows a nonlinear path, resulting in twisting or rotation of the brain within the skull (see Figure 20-9C). Rotation forces produce compressive, tensile, and shearing strains that cause distortion and injury of involved brain tissues and possible vascular disruption. Maximal stress from rotational forces is directed toward areas where tissues with different densities interface (e.g., between cerebral and fibrous tissue, between gray and white matter). The degree of injury from these rotational forces depends on the extent, rate, and direction of angular acceleration.11,12 Motor vehicle crashes are a common cause of this type of mechanism.
Penetration
A gunshot may create a single injury tract or, when a low-velocity missile is involved, it may ricochet within the cranial cavity and create multiple destructive missile tracts within the brain. A bullet projected through the skull and brain creates shock waves and a cavity that may expand to many times the bullet’s diameter and then collapse, causing extensive local and remote tissue damage. High-velocity bullets that strike with much energy tend to cause greater tissue destruction from the temporary cavitation and shock waves than do low-velocity missiles, which wound primarily by direct tissue destruction.13 Death associated with firearm-related TBI is the result of extensive structural damage and uncontrolled intracranial hypertension associated with intracranial hemorrhage and extensive cerebral edema. Patients may remain conscious immediately after gunshot wounds to the head, only to deteriorate rapidly with the onset of edema around the missile tract.
PATHOPHYSIOLOGY OF TRAUMATIC BRAIN INJURY
ASSOCIATED SCALP AND SKULL INJURIES
Skull Fractures
Impact to the head can fracture the skull, which may or may not be accompanied by injury to the underlying brain. The presence of skull fractures has been identified as a significant predictor for injury-related abnormalities on computed tomography (CT) scan even after minor TBI.14,15 The degree of skull injury depends on the mass, characteristics (e.g., shape), velocity, momentum, and direction of the object that impacts the head and the thickness of the skull at the point of contact.
Depressed fractures are the result of the cranial bone being forced below the line of the normal skull contour. These fractures typically result from high-velocity contact sustained over a small surface area. The fragmented bone particles may be embedded into the brain tissue, resulting in cortical laceration and hemorrhage. A depressed skull fracture associated with a scalp laceration is called a compound or open fracture. These injuries are associated with an increased incidence of posttraumatic seizures and a high risk for intracranial infection.14 Treatment of these open fractures, particularly if the fractured bone is depressed greater than the cranial thickness, usually consists of surgical debridement, evacuation of clots and bone fragments, elevation of the depressed bone, and repair of the lacerated dura as soon as possible after injury.14 Closed depressed skull fractures may not require surgical intervention.14
Basilar skull fractures are located at the base of the cranium. The five bones that form the skull base are the occipital bone, the cribriform plate of the ethmoid bone, the orbital plate of the frontal bone, the sphenoid bone, and the petrous and squamous portions of the temporal bone (Figure 20-10). Basilar skull fractures are most common in the anterior and middle fossae. Fractures of the cranium base may be linear, compound, or depressed; most commonly they are linear fractures that extend downward into the base of the skull. Basilar skull fractures can be difficult to detect with routine radiographic studies. Clinical examination findings typically identify their presence. Common symptoms of an anterior fossa fracture include periorbital ecchymosis (raccoon’s eyes), rhinorrhea (CSF or blood draining from the nose), and conjunctival hemorrhage without evidence of direct ocular trauma. Physical findings common in the presence of a middle fossa fracture include mastoid ecchymosis (Battle’s sign), otorrhea (CSF or blood from the ears), hemotympanum, facial nerve paralysis, and hearing loss.
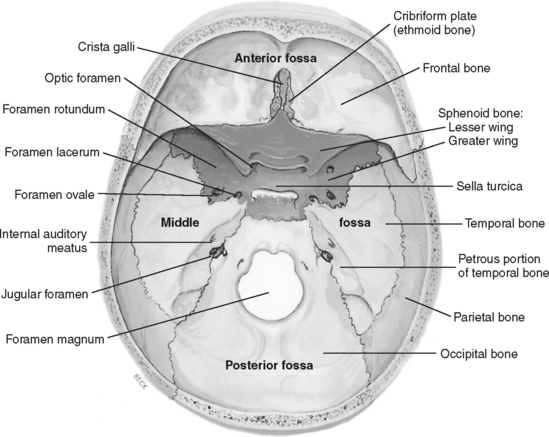
FIGURE 20-10 Base of the skull showing the cranial fossae.
(From Slazinski T, Littlejohns LR: Anatomy of the nervous system. In Bader MK, Littlejohns LR, editors: AANN core curriculum for neuroscience nursing, ed 4, Philadelphia, 2004, W. B. Saunders, p. 32. Modified from Thibodeau GA, Patton KT: Anatomy & physiology, ed 5, St. Louis, 2003, Mosby.
The presence of CSF otorrhea or rhinorrhea indicates not only that a basilar skull fracture exists, but also that the dura is lacerated, which substantially increases the risk for intracranial infection. The CSF should be permitted to drain freely by only applying a loose sterile dressing over the drainage site. Medical management typically includes observation, bed rest, head of bed elevation and precautions to prevent worsening the leak (e.g., do not blow the nose, avoid Valsalva maneuver).16 If the CSF leak persists, a lumbar drain may be placed for a few days to divert CSF and encourage closure of the dural laceration. Although more than 80% of CSF fluid leaks stop spontaneously, those that persist after 7 to 14 days may require surgical dural repair.16
TYPE OF BRAIN INJURIES
Brain injuries can be classified as focal or diffuse. Focal brain injuries occur in a localized region, and diffuse brain injuries involve a large generalized area. Many patients with TBI, especially severe injury, have a combination of focal and diffuse injuries.11
Focal Brain Injuries
Cerebral Contusions.
Contusions are bruises of the brain tissue with associated hemorrhage and edema formation that may lead to subsequent tissue necrosis and infarction. Surrounding the contusion is a zone of edema and low cerebral blood flow that may be at high risk for ischemic insult.17 Unlike the severely ischemic center of the contusion, the pericontusional zone or penumbra may eventually regain regional blood flow.17
Contusions constitute the most common type of brain injury. These lesions may occur beneath an area of skull deformation or secondary to acceleration-deceleration, rotation, or penetrating mechanisms of injury. Although contusions may occur anywhere in the brain, the most common sites are the frontal and temporal lobes.11,17,18 Movement restrictions created by the cranial walls, dural folds, and irregular bony projections on the basilar skull surface in the frontotemporal regions explain the prevalence of brain injury in these areas. Contusions are most often multiple and frequently occur in association with other lesions.
Signs, symptoms, and severity of the injury depend on the site and extent of the contused brain. Isolated contusions generally do not produce immediate loss of consciousness. When coma is present, it is usually the result of an associated diffuse injury. Because the majority of contusions occur in the frontal and temporal lobes, patients usually present with localizing personality, memory, executive function, behavior, motor, and language deficits.18 Progressive hemorrhage and edema formation can cause an abrupt mass effect, resulting in ICP elevation, brain compression, and, eventually, herniation.
Epidural Hematoma.
An epidural hematoma (EDH), also known as an extradural hematoma, is an accumulation of blood between the skull and dura mater (Figure 20-11). Epidural hematomas are most commonly located in the temporal region. These hematomas are often associated with a linear fracture of the thin temporal bone that lacerates the underlying middle meningeal artery, causing an accumulation of blood in the extradural space. Less frequently, the source of hemorrhage is venous (e.g., diploic veins, venous sinuses, middle meningeal vein).19 As the hematoma accumulates, it strips the dura from the inner table of the skull and compresses the underlying brain. This local mass effect eventually causes brain herniation and can lead to death.
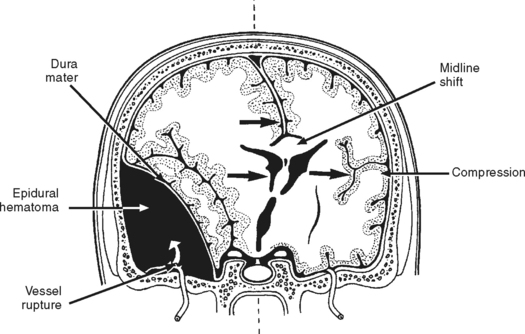
Signs and symptoms of EDH depend on the source and rate of blood accumulation. Clinical manifestations are usually seen within 6 hours of injury. The classic clinical presentation of a patient with an EDH is described as a period of unconsciousness believed to occur because of a concussion, followed by a lucid period of variable length, after which the level of consciousness deteriorates as the hematoma expands. The lucid interval is not pathognomonic of EDH, as it reportedly occurs in less than half of patients with epidural hematomas.19 Other common manifestations of an expanding hematoma are pupil abnormalities (e.g., unilateral and eventual bilateral pupil dilation and decreased reactivity to light) and altered motor function (e.g., hemiparesis or hemiplegia, flexion or extension posturing).
Any EDH creating a significant mass effect requires immediate surgical evacuation and ligation of bleeding vessels. The evidenced-based Guidelines for the Surgical Management of Traumatic Brain Injury recommends surgical evacuation of an EDH if the size exceeds 30 cm3 regardless of the GCS score. Nonoperative management with follow-up CT scans and close observation of neurological status can be considered if the EDH is less than 30 cm3 with less than a 5-mm midline shift and less than 15-mm thickness in patients with a GCS score more than 8 without any focal symptomatology. In comatose patients with a GCS score of 8 or less and unequal pupil size, surgical evacuation of the EDH is strongly recommended as soon as possible.19 If this is not possible and rapid neurological deterioration occurs, a burr hole may be placed ipsilateral to the dilated pupil or contralateral to the motor deficits to evacuate the hematoma. Although significant brain injury may result from brain compression by the hematoma, there typically is little or no underlying primary brain damage. Therefore, early diagnosis and treatment may yield a good prognosis.
Subdural Hematoma.
Subdural hematoma (SDH) is a collection of blood beneath the dura mater and above the arachnoid layer of the meninges (Figure 20-12). These lesions are usually caused by rapid acceleration-deceleration mechanisms that tear veins bridging the subdural space, where they connect the brain’s cortical surface to the dural sinuses. Rupture of small cortical arteries may also be the source of subdural hemorrhage, which may extend over the entire hemisphere.
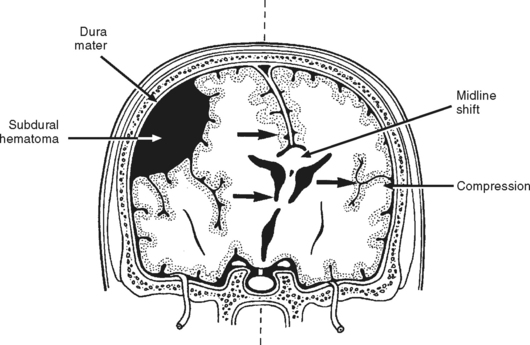
Patients with acute SDHs have one of the highest reported mortality rates of all types of TBI, averaging around 40% to 60%.20 High mortality associated with acute SDH is believed to be associated with the large amount of primary brain damage (i.e., contusion, laceration, and vascular disruption) underlying the clot, as well as the brain compression that occurs as the lesion expands. The length of time from clinical deterioration or brain herniation to surgical intervention for SDH is significantly related to outcome.20,21 Even with prompt surgical removal of the SDH, development of contusion, intracerebral hemorrhage or infarction of brain underlying the evacuated clot could increase mortality.
Acute SDH creating a mass effect requires rapid clot evacuation, hemorrhage control, and resection of underlying nonviable brain. The Guidelines for the Surgical Management of Traumatic Brain Injury recommend evacuation of a clot with a thickness greater than 10 mm or causing a midline shift of greater than 5 mm regardless of GCS score.20 Patients with an SDH less than 10 mm thick, less than a 5 mm midline shift, and a GCS score less than 9 should undergo surgical removal of the clot if the GCS score declines by more than 2 points or the ICP is greater than 20 mm Hg, or the pupils are fixed and dilated or assymetrical.20 Surgical evacuation of an SDH should occur as soon as possible, because data suggest those who undergo surgical SDH evacuation within 2 to 4 hours have better outcomes than those who undergo delayed operative intervention.20 The operative procedure generally involves initial trephines or burr holes to locate the clot, followed by conversion to a craniotomy flap for optimal evacuation. A soft drain may be left in the subdural space for 24 to 48 hours. Postoperative complications may include brain swelling with subsequent increased intracranial pressure, clot reaccumulation, delayed intracerebral hemorrhage, and seizures. Small acute SDHs producing no significant mass effect may not require surgical intervention and are absorbed spontaneously.
Subacute SDHs have similar but slower developing symptomatology than that seen with acute SDHs. Symptoms of a subacute SDH become apparent 2 days to 2 weeks after head injury. These symptoms tend to be more benign and the prognosis is generally better than with acute subdural hematoma. The improved prognosis is related to less severe underlying brain contusion and the decreased likelihood that the patient will deteriorate to the point of brainstem compression. Like acute SDH, subacute SDH typically requires surgical evacuation.
Intracerebral Hematoma.
An intracerebral hematoma (ICH) is a well-defined clot located within the brain parenchyma that, like a contusion, is typically surrounded by an area of edema and low blood flow17 (Figure 20-13). Most likely the bleeding originates from cerebral vessels that rupture at the time of injury. Similar to contusions, most ICHs occur in the frontal and temporal lobes and, less frequently, deep within the cerebral hemispheres or in the cerebellum.17 ICHs may be multiple and associated with other lesions, particularly contusions. When an ICH is in continuity with a SDH, it is known as a burst lobe. Evidence of an isolated ICH after head trauma should cause health care providers to consider if a hypertensive bleed may be the source of the hemorrhage.
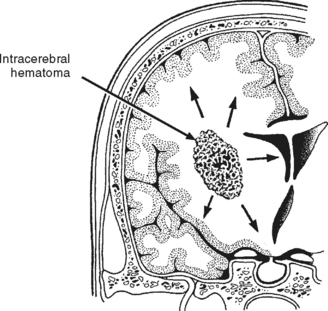
ICHs are complicated by aggressive focal edema and increasing mass effect, which leads to elevations in ICP and further neurologic deterioration. Deterioration can occur as late as 7 to 10 days after injury, although the majority of neurologic deterioration typically occurs in the first 48 to 72 hours.17 The initial size of the hematoma is a predictor for subsequent lesion expansion with larger lesions demonstrating greater likelihood of enlarging than smaller ones.17 Surgical evacuation of the ICH is not always beneficial or possible.
Subarachnoid Hemorrhage.
Signs and symptoms of SAH include decreased level of consciousness, motor deficits (e.g., hemiparesis), pupil abnormalities, and possibly evidence of meningeal irritation, such as headache, photophobia, and nuchal rigidity. Potential complications of SAH include ICP elevation, posttraumatic hydrocephalus, and cerebral vasospasm. Patients demonstrating SAH on CT scan tend to have a significantly worse outcome than those without evidence of SAH.17 Treatment for SAH includes placement of an intraventricular catheter to drain bloody CSF and, if necessary, management of intracranial pressure.
Diffuse Brain Injury
Unlike focal injuries, diffuse injuries do not consist of damage to a single localized area of the brain; instead, damage is more widespread. Acceleration-deceleration and rotational forces, usually associated with motor vehicle crashes but sometimes falls, assaults, or other types of trauma, are the most common mechanisms that initiate the pathophysiologic process that results in diffuse injuries to the brain.11,12 These forces create tension and compression on, and shearing of, nerve fibers, resulting in variable amounts of axonal damage. The magnitude and rate of strain and direction of the force during brain trauma determine the severity of axonal injury.11,12 Diffuse axonal injury (DAI), also known as traumatic axonal injury, creates an injury pattern that tends to primarily affect the subcortical white matter throughout the hemispheres, especially at the gray-white matter interface, the corpus callosum, and the brainstem (Figure 20-14).12,22–24
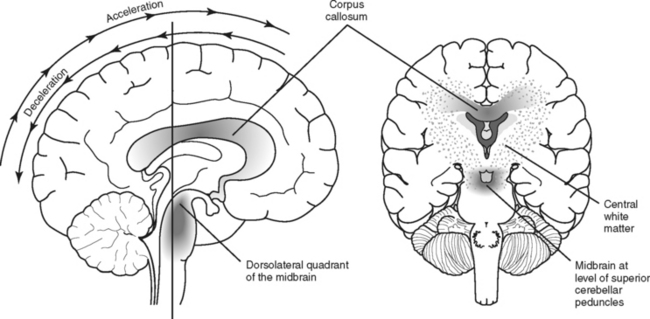
Although in the case of severe TBI, a portion of axons may be disrupted physically (axonotomy) at the time of injury, more often with DAI, axons remain structurally intact but may be functionally impaired. Axons that retain structural integrity but sustain internal damage and functional disruption may recover; experimental models suggest they may suffer delayed or secondary axonotomy hours after the injury.11 In other words, all injured axons are not transected at the time the brain is subjected to inertial forces; rather, neuronal pathology associated with DAI is a complex progressive event (described in greater detail in a later section on secondary injury).11,18 The amount, severity, and location of axonal damage will determine the clinical severity and outcome of diffuse brain injury.18,25 Diffuse brain injuries lie on a continuum of severity ranging from the mildest form of DAI, concussion, with little or no sustained brain dysfunction, to moderate and severe diffuse axonal injury that may produce disabling deficits or death.11,18,22
Patients with DAI typically progress through three phases of recovery.18 The first phase is immediate unconsciousness, which may not occur in the mildest cases of DAI. In the next longer lasting phase, the patient regains consciousness and is confused, which is linked to anterograde amnesia.18 In the even lengthier final phase, the patient undergoes restoration of cognitive function. Severity of DAI correlates with the duration of each phase and degree of impairment associated with the stages.18 With mild DAI, the duration of each phase is fairly short with few if any lasting impairments, whereas the opposite is true with severe DAI.18
Mild Diffuse Axonal Injury: Concussion.
Concussion is a transient disturbance of neurological function. Axonal shear, tension, or strain occurs, resulting in conduction impedance and transient loss of function. No anatomical evidence of brain injury is apparent on CT scan, although magnetization transfer magnetic resonance imaging (MRI), diffusion weighted MRI,23 diffusion tensor MRI (DTI),26 magnetic resonance spectroscopy (MRS),27 functional MRI (fMRI), positron emission tomography (PET), and single-photon emission computed tomography (SPECT) scans,28 though not routinely performed, are more frequently able to identify abnormalities after this type of axonal injury.22,29 The diagnosis is based on the patient’s history, clinical neurologic findings, and a CT scan that rules out other intracranial lesions.
Manifestation of a concussion may include somatic, cognitive, or psychological impairments. A temporary loss of consciousness, usually lasting only a few minutes, may or may not occur. Patients may experience retrograde amnesia (inability to recall events just preceding the injury) or posttraumatic amnesia. The duration of amnesia may predict the severity of symptoms and deficits (e.g., depression, dizziness, cognitive impairments) associated with concussion and is considered a good measure of the injury severity.30,31 Signs and symptoms also may include headaches, confusion, dizziness, vertigo, nausea, visual disturbances, subtle personality changes, depression, difficulty in concentration, decreased information-processing speed, poor memory, behavioral disorders, emotional lability, apathy, irritability, fatigue, increased anxiety, and insomnia.30,31 These manifestations, which can vary considerably among patients and may persist for days, weeks, or even months after the concussion, are collectively known as postconcussive syndrome (PCS). Persistence of these problems may be related to the degree of neuronal damage, emotional distress (e.g., anxiety or depression), personal set factors (i.e., older age, female gender), motivational issues, cognitive factors, or a combination of factors.29,32
Concussions are very common but, because of their apparently benign nature, medical attention is often not sought or there is a delay in seeking treatment for persistent symptoms. Patients who do seek medical care for an isolated concussion usually are not admitted to the hospital and receive no follow-up treatment at outpatient facilities. Patients and their families should receive information and supportive counseling on PCS, especially about the potential difficulties that may become evident after return to home, work, or school. Patients with persistent or disabling postconcussive symptoms may benefit from referral to a specialist (e.g., a neuropsychologist) for more extensive assessment and treatment. Patients should be advised to avoid activities (e.g., contact sports) that present a high risk for repeat concussion during their recovery period, because another concussion before resolution of the initial injury may cause cerebral autoregulatory dysfunction, progressive cerebral edema, neurological deterioration, and possibly death.33 The reader is referred to expert consensus recommendations and reviews on this topic to acquire more thorough information regarding return to play recommendations for athletes who sustain a concussion.30,34
Moderate to Severe DAI.
Tension, shearing, and compression strains created by rotational and acceleration-deceleration forces cause widespread axonal cytoskeletal and functional disruption in the cerebral hemispheres, the corpus callosum, the brainstem, and, less commonly, the cerebellum. When DAI is more severe, the duration of the recovery phases (i.e., unconsciousness, emerging consciousness, and postconfusional restoration of cognitive function) is prolonged and plagued with more significant and sometimes lasting impairments. Those with the most severe DAI may halt progression through the stages of recovery.18
As severity of DAI increases, coma may last more than 24 hours. Confusion and long-lasting amnesia typically follow coma emergence. Most patients with moderate DAI move purposefully or withdraw from pain, but some may exhibit transient decorticate or decerebrate posturing. Mild to severe attention, behavioral, cognitive, memory, and/or executive (e.g., reasoning, speed of processing information) deficits usually persist.25 Memory and executive dysfunction are the most common neuropsychological impairments found in patients that had CT scan or MRI findings compatible with DAI.25
Severe diffuse axonal injury results in extensive anatomic and functional disruption of axonal white matter fibers. Unlike less severe cases of DAI in which typically there is no visible brain injury on CT scan, in severe DAI, small tissue and vessel tears may be apparent. These tears appear as small focal lesions typically located in the hemispheric subcortical lobar white matter, corpus callosum, basal ganglia, brainstem and sometimes the cerebellum.12,23 Involvement of the corpus callosum and dorsolateral aspect of the upper brainstem corresponds with greater severity of DAI and poorer outcome.18 Because these lesions are small, they may be missed or difficult to appreciate on initial CT scan, and early resolution makes detection difficult in subsequent scans. MRI is more effective than CT in visualizing small lesions associated with DAI, including lesions deep in the white matter of the cerebral hemispheres.18,22 Diffuse cerebral edema also may be seen with severe diffuse axonal injury. The extent of DAI, particularly when not severe, may be better visualized through other diagnostic technologies not typically employed in assessment of TBI as previously mentioned under the section on concussion. Extensive neuronal loss eventually results in brain shrinkage. Follow-up CT scans typically show cerebral atrophy, as evidenced by enlarged sulci and ventricular dilation.
Clinically, severe DAI is manifested by deep, prolonged coma lasting days to months. Signs of severe cerebral and brainstem dysfunction, including decorticate and decerebrate posturing, are often present on admission or develop with 24 hours of injury. Diencephalic involvement usually results in onset of tachycardia, systemic hypertension, tachypnea, hyperpyrexia, and profuse diaphoresis (hyperhidrosis) covering the face and, less frequently, the neck and upper thorax accompanied by increased muscle tone and abnormal posturing.35 It is theorized that this syndrome, called neurosweats, diencephalic fits, sympathetic storming, paroxysmal autonomic instability with dystonia, or dysautonomia, is caused by dysfunction of the autonomic control centers (hypothalamus or thalamus) located in the diencephalic region or disruption of connections to these centers from other areas of the brain.35 Diencephalic signs and abnormal posturing usually resolve a few weeks after injury, but may persist for months.35
Because of the extensive structural damage to neurons, severe DAI has a high mortality. The majority of severe DAI survivors typically have major residual disabilities that may not be evident until consciousness improves. Major sequelae of severe DAI may include cranial nerve dysfunction, sensorimotor impairments (e.g., paresis, tremors, spasticity, dyspraxia, problems with speech) and, most commonly, significant deficits in cognition and neuropsychological capability.12 Severe DAI is the most common cause of major disability and persistent vegetative state after TBI.36






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


