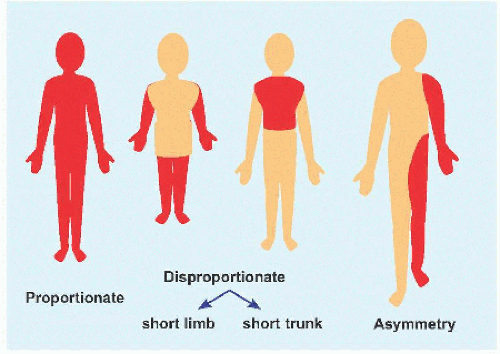
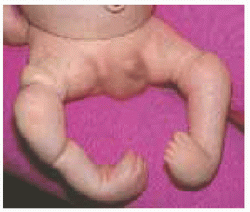
A Short stature This may proportionate or disproportionate, or it may affect a region of the body asymmetrically.
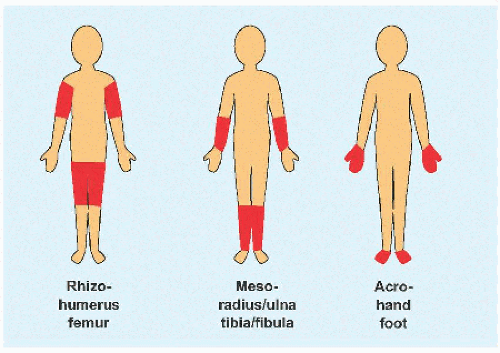
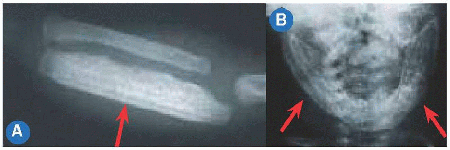
B Micromelia Shortening may affect the “root” or proximal segment, the “middle” segment, or the “tip” of the limb.
Syndrome
Facies
Achondroplasia
Frontal bossing, depressed nasal bridge
Apert
Proptosis, downsloping palpebral fissures
Camptodactyly
Flat face
Cantu syndrome
Full lips, long philtrum
Cornelia de Lange
Synophrys, “carp” mouth
de Barsy
Progeroid
Down
Mongoloid
Emanuel
Low-hanging columella
Freeman-Sheldon
Whistling face
Hurler
Gargoyle
Larsen
Dish face
Pseudoachondroplasia
Normal face
Proteus
Elephant man
Rubinstein-Taybi
Squinting smile
Silver-Russell
Triangular face
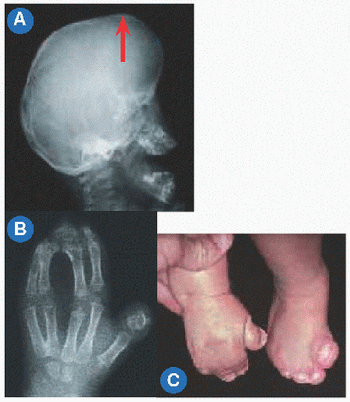
C Facies Many syndromes may be distinguished by appearance of the face.
GENERAL CONSIDERATIONS
The title syndromes is used for lack of a more inclusive term. Syndromes are arranged in alphabetical order for simplicity. There is no comprehensive system of classification that is complete, because of diverse causes, heterogeneity of presentation, evolution of expression with growth, as well as rapid and continual medical discovery. These are diseases first and orthopedic problems second: most important is evaluation, because orthopedic management will not solve the primary problem and may deliver a fair outcome at best. This contrasts with disorders of the musculoskeletal system that occur in an otherwise normal child, where orthopedic management is the focus. All tissues of the skeleton may be affected, from bone to cartilage to surrounding soft tissues. The clinical presentation is broad, from premature osteoporotic fracture in the adult to perinatal lethal. While individually the disorders may be rare, collectively, their incidence may be as high as 1/5,000 births. It is important to realize that many diagnoses do not represent a single disease but rather a heterogeneous group of disorders of which only some subtypes are well characterized while others originate in single case reports.
Language
Syndrome, used by Galen as a compound of Greek óõí: “with, together” and δροµοσ: “a course, race, running,” signifies “a concurrence” of signs in, or the clinical presentation of, a disease.
Short stature may be defined as <2 standard deviations below mean height or below 2.5 percentile. An alternative guideline is height <5 ft. (150 cm). Short stature may be divided into proportionate, affecting the entire body equally, or disproportionate. Terms such as midget for the former and dwarf for the latter are not universally accepted. Little person is neutral and is aligned with the Little People of America.
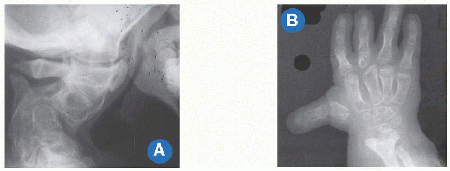
Disproportion of short stature may arise from the limbs, referred to as micromelia (Greek µικροζ: “small” and µελοζ: “limb”). Disproportionate shortening of the “trunk” is known as microcormia (Greek κορµοζ) [A]. Limb shortening may be asymmetric. Alternatively, limbs may be short at the “root,” known as rhizomicromelia (Greek ρι§α); at the “middle” segment, known as mesomicromelia (Greek µεσοζ); or at the “tip,” known as acromicromelia (Greek ακροζ) [B]]. This terminology comes from radiographic classification based upon the region of bone principally affected, such as epiphysis versus metaphysis versus diaphysis. The convenience of this system has led to its wide adoption; however, it is simplistic, bears no relationship to morbidity, and frequently suggests a connection between entities where there is none.
Of the skeleton, dysplasia (Greek δvζ: “bad” and πλασσω: “I form”) represents a generalized affection of the skeleton. Dysostosis (Greek οστεοv: “bone”) refers to involvement of a single bone or a group of physically or functionally related bones. Dysmorphism (Greek Morpheus, God of Sleep who may take any human “form” in dreams) is applied to a “bad form” of body part, often the facies (Latin) or “face” that can distinguish a specific disorder [C].
Classification
These disorders have been given descriptive names according to clinical presentation, pathogenesis, or radiographic appearance. Achondroplasia emphasizes cartilage as locus of disease, while osteogenesis imperfecta distinguishes bone as the abnormal tissue. Other skeletal dysplasia may be grouped according to whether they affect the metaphysis or the epiphysis of a long bone. Identification of genetic mutations has allowed molecular typing. For example, the type II collagenopathies span a spectrum from the severe Kniest dysplasia to spondyloepiphysial dysplasia and Stickler disease to the relatively mild precocious osteoarthritis. Challenges of molecular classification include:
The molecular defects are numerous and evolving.
Clinically unrelated disorders may have the same molecular defect. Achondroplasia is caused by mutation of one type (3) of fibroblast growth factor receptor, while other types produce the craniosynostosis syndromes of Pfeiffer (1) and Crouzon (2).
Clinically related disorders may have different molecular defects. Mutations in the gene encoding type IX collagen and the gene encoding cartilage oligomeric matrix protein both have been found in multiple epiphysial dysplasia.
The molecular defects are heterogeneous, obscuring the biologic pathways to disease. The highest expression of fibroblast growth factor receptor 3 is in the brain, yet it is the expression in cartilage during endochondral ossification that is responsible for the clinical manifestations of achondroplasia.
Despite these limitations, knowledge of molecular biology may aid understanding of the tissue distribution of disease. Type I collagen is the principal structural protein of bone, dentin, and sclera, hence the association (and clinical subclassification) of osteogenesis imperfecta with dentinogenesis imperfecta and blue sclerae. Type II collagen occurs in cartilage and vitreous humor. As a result, Stickler arthroophthalmopathy may include osteoarthritis and retinal detachment.
Evaluation
Assess clinical features, support with imaging, and verify with histologic analysis and laboratory studies if available.
Stature After facial appearance, stature is most distinguishing. Stature may be short, normal, or tall. Short stature may be proportionate, such as in endocrinopathy, or disproportionate, such as skeletal dysplasias. Few syndromes feature tall stature, such as Marfan. Neuromuscular disorders do not affect stature significantly.
Family history Many syndromes are inheritable. They may be autosomal or X-linked and dominant or recessive. Others represent new spontaneous mutations. Neurofibromatosis type 1 is transmitted as an autosomal dominant to half affected children, while in the other half, the disorder arises spontaneously.
Development Some syndromes are characterized by global delay, such as Prader-Willi syndrome. Others affect cognitive or motor function separately, such as muscular dystrophy. Developmental delay may be temporal or permanent; for example, in achondroplasia, motor development may be delayed in the first 2 years, due to macrocephaly and ligamentous laxity.
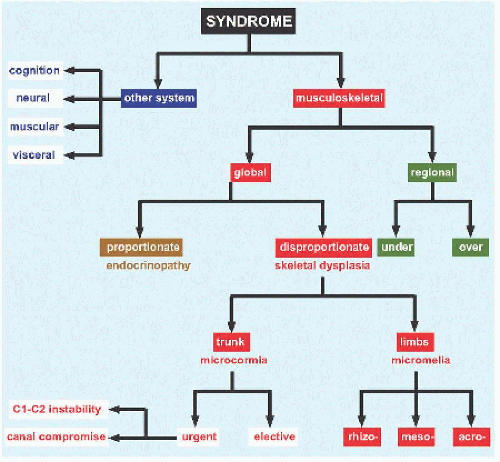
System Divide conditions into those that affect the musculoskeletal system and those that affect other systems and viscera [D]. Within the musculoskeletal system, conditions may be categorized geographically according to the part affected.
Pathognomonic features From Greek παθοζ: “disease” and γιγvωσκω: “I know,” these may form the nidus around which other findings may be assembled. This approach represents a primary focus on the disease, in contrast with the system approach, which focuses on the manifestation [E].
Imaging
RÖNTGENOGRAMMES These form the basis of evaluation. They may describe a disease geographically.
Part of the skeleton, for example, spondylo- for spine
Part of a long bone, for example, epiphysial, metaphysial, and diaphysial
Röntgenogrammes may describe the skeleton qualitatively.
Increased density, for example, osteopetrosis
Reduced density, for example, osteogenesis imperfecta
Mixed, for example, stippling of epiphysis in chondrodysplasia punctata
Röntgenogrammes may reveal features that, while not always present in every patient, are pathognomonic [E].
Radiographic features vary according to age. Many features that may aid diagnosis in childhood disappear by adulthood, such as epiphysial stippling. These features are considered dynamic, a manifestation of abnormal bone development.
Sandwich vertebrae, rugger jersey spine, endobones.
Osteopetrosis (of Albers-Schönberg)
Tree frog feet.
otopalatodigital
Secondary ossification center at base of second metacarpal and metatarsal
Second metacarpal pseudo-epiphysis
Silver-Russell
E Pathognomonic features These may offer direction in a sea of complexity.
ULTRASONOGRAPHY This modality may establish a prenatal diagnosis. The fundamental finding is short limbs for gestational age. A small thorax (including reduced thoracic:abdominal ratio) is a feature of lethal skeletal dysplasias. There may be clinical expression of osseous disease, for example, fractures in osteogenesis imperfecta. There may be delayed or absent ossification, for example, of the clavicles in cleidocranial dysplasia. There may be regional abnormalities, for example, clubfoot in diastrophic dysplasia.
OTHER IMAGING MODALITIES These are employed according to specific relevance to disease. In achondroplasia, foramen magnum stenosis may be measured on computed axial tomography. Published disease-specific standards are available based upon such measurements. Critical stenosis of the vertebral canal due to spine deformity may be evaluated by magnetic resonance imaging.
Laboratory analysis This most commonly is performed of blood, urine, and skin. Chromosome number, size, position, and staining pattern may be determined by karyotyping blood. Hormone serum levels, for example, growth hormone, thyroxine, and thyroid stimulating hormone, identify endocrinopathy. The mucopolysaccharidoses are characterized by enzyme deficiency leading to reduction of product and accumulation of precursor that is detected by urine assay. The clinical diagnosis of Marfan syndrome may be confirmed by immunohistochemical staining or pulse-chase analysis of fibrillin-1 protein in cultured skin cells obtained from skin biopsy. Biochemical and molecular analysis of skin and blood for type I collagen mutation aids the diagnosis and typing of osteogenesis imperfecta. Ehlers-Danlos VI may be confirmed by insufficiency of hydroxylysine on analysis of hydrolyzed dermis, reduced lysyl hydroxylase activity in cultured skin fibroblasts, and altered ratio of lysyl pyridinoline to hydroxylysyl pyridinoline in urine.
Algorithm
The following is a singular and simplified approach to turn the key and open the door to this multidisciplinary area into which may be drawn the orthopedic surgeon at times insecure and intimidated [F].
F A simple algorithm for initial navigation of a syndromic patient.
ACHONDROPLASIA
Most common skeletal dysplasia, with incidence ˜ 1:25,000 live births.
Gain-of-function fibroblast growth factor receptor 3 (FGFR3) mutation on chromosome 4p16.3 codon 380 glycine. Chondrocyte activation of FGFR3 increases bone formation and accelerates fusion of ossification centers with premature synchondrosis closure, limiting endochondral bone growth.
This may be transmitted as autosomal dominant with complete penetrance; however, most are new spontaneous mutations. A family of FGFR3 mutations is recognized ranging from mild (hypochondroplasia) to moderate (achondroplasia) to severe (thanatophoric dysplasia).
Features do not become apparent on ultrasonogram until after 16 weeks.
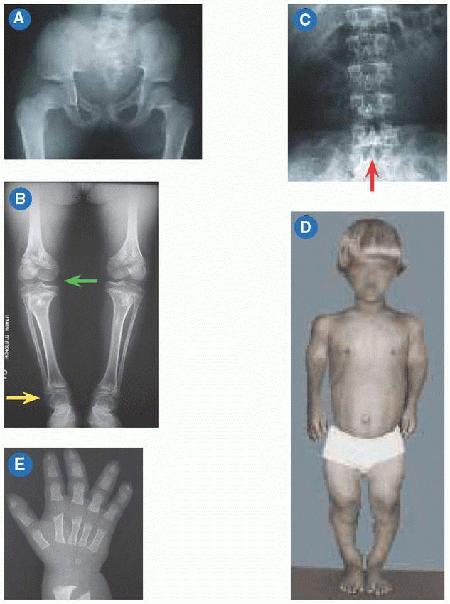
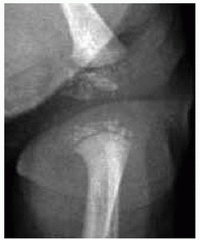
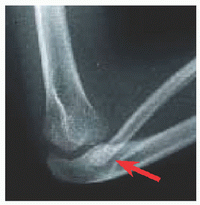
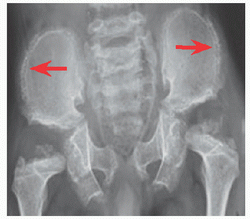
Abnormal endochondral with spared membranous ossification produces a large head with narrow foramen magnum, “champagne pelvis” with constricted triradiate cartilage [A], long clavicles with broad shoulders, long fibulae with varus ankles and knees [B], and short pedicles with lumbar spinal stenosis [C]. Hypotonia in infancy, rhizomicromelia [D], frontal bossing with midface hypoplasia, trident hand [E], thoracolumbar kyphosis, joint contractures (including at the hips exaggerating lumbar lordosis), and “chevron” metaphysis.
Decompression of brainstem for foramen magnum stenosis, based upon MRI compared with normative data, somatosensory evoked potentials, and polysomnography. Spine osteotomy and fusion for thoracolumbar kyphosis. Due to pedicle hypoplasia, decompression for lumbar spinal stenosis must include articular process excision, requiring fusion and instrumentation. Osteotomy for genua vara. Physiodesis of distal fibula for ankle varus.
Nonsurgical treatment includes weight control, management of frequent middle ear infections and dental crowding, adenotonsillectomy and nasal mask continuous positive airway pressure for obstructive sleep apnea.
Pharmacotherapy includes growth hormone (although there is no consensus), and BMN-111, a stabilized version of C-type natriuretic peptide that inhibits the FGFR3 pathway.
Future strategies for treatment include the following:
Chemical inhibitors of FGFR3 tyrosine kinase
Antibodies to interfere with binding of FGF ligands to FGFR3
C-type natriuretic peptide antagonism of FGFR3 downstream signaling by inhibition of mitogen-activated protein kinase pathway in growth plate chondrocytes, thereby recovering bone growth
ACRODYSOSTOSIS
Type 1 caused by heterozygous mutation in PRKAR1A gene on chromosome 17q24, and type 2 by mutation in PDE4D gene on chromosome 5q12.
Peripheral dysostosis (short tubular bones) of hands and feet. Reduced interpedicular distance producing spinal stenosis. Stippling of coneshaped epiphysis resolves spontaneously in the first few years of age.
AMNIOTIC BAND SYNDROME
Also known as Streeter anomaly.
Herniation of members through ruptured amnion results in constriction, vascular occlusion, and necrosis. ADAM (amniotic deformity, adhesions, mutilations) complex includes associated terminal transverse defects and cleft lip and palate. LBWD (limb body wall defect) includes associated body wall and visceral defects explained by pressure on the embryo during the first 4 weeks.
AMYOPLASIA
See arthrogryposis.
ANTLEY-BIXLER SYNDROME
Mutation of cytochrome P450 oxidoreductase on 7q11.23.
Trapezoidocephaly secondary to lambdoid and coronal synostosis, radiohumeral and radioulnar synostosis, and camptodactyly.
Abnormal steroidogenesis and genitourinary and cardiac anomalies.
Airway obstruction may lead to demise in the neonatal period.
APERT SYNDROME
Mutation in fibroblast growth factor receptor 2 gene (FGFR2). By contrast with dominant mutations in the FGFR3 gene, which affect endochondral ossification resulting in achondroplasia, dominant mutations of FGFR1 and FGFR2 cause the craniosynostosis syndromes of Apert, Crouzon, and Pfeiffer, which involve bones arising by membranous ossification.
Autosomal dominant; however, most de novo mutations.
Wheaton first reported two cases of what Apert called acrocephalosyndactyly. Acrocephaly is due to coronal synostosis [A]: early decompressive craniectomy may limit mental deficiency. Syndactyly and synonychia produce “mitten” hand [B] and “sock” foot [C]: the former is an indication for release, and the latter for osteotomy or ostectomy to relieve pressure. Failure of cervical segmentation, broad thumbs and halluces, and carpal and tarsal coalition. Cardiac, respiratory, nervous, abdominal, and genitourinary anomalies.
ARTHROGRYPOSIS
Originally termed arthrogryposis multiplex congenita. Arthrogryposis refers to a heterogeneous group of disorders of which the common feature is multiple congenital joint contractures. Fetal akinesia due to maternal disease, intrauterine constraint, or vascular compromise retards development of nerves, muscles, or connective tissues. The earlier and longer the loss of movement, the more severe the deformities. Muscle is replaced by fibrofatty tissue. Arthrogryposis without other system disease is subclassified as amyoplasia. Distal arthrogryposis, affecting hands and feet, is autosomal dominant. Intelligence is normal. Limited joint motion, medial rotation of shoulders, extension of elbows, flexion and ulnar deviation of wrists, camptodactyly, thumb in palm, hip dislocation, knee contracture, clubfoot, and scoliosis. Loss of cutaneous creases with joint dimpling. Operative reduction of dislocated hips is controversial. Femoral shortening facilitates treatment of flexion contracture. Center the arc of motion at 15 degrees: walking is easier on straight knees. Talectomy may be necessary for clubfoot. Stiffness limits correction of scoliosis.
BEALS SYNDROME
Type
Features
I
Known as auriculo-osteodysplasia. Autosomal dominant.
Short stature, auricular anomalies including elongation of lobe with secondary posterior lobule, radiocapitular dysplasia with head of radius dislocation, hip dysplasia.
II
Known as congenital contractural arachnodactyly. Mutation in fibrillin-2 gene at 5q23-q31.
Marfan syndrome without visceral involvement and with crumpled ear helices as the hallmark, due to expression of fibrillin-2 in auricular cartilage.
BECKWITH-WIEDEMANN SYNDROME
Mutations in several imprinted genes within 11p15.5 region, as well as mutation of 5q35. The former also is affected in Silver-Russell syndrome, while the latter in Sotos syndrome.
Autosomal dominant with variable expressivity, as well as de novo mutation.
Overgrowth, including macroglossia, exophthalmos, limb hypertrophy, and visceromegaly. Tumor diathesis, including Wilms tumor, hepatoblastoma, neuroblastoma, and adrenal carcinoma. Posterior helical ear pits, abdominal wall defects including umbilical hernia, and renal anomalies. Neonatal hypoglycemia and history of hydramnios and prematurity.
BRACHYDACTYLY
Multiple mutations identified for different types and subtypes. Classified into groups A to E, each of which are subclassified. First syndrome in humans in which Mendelian inheritance was described.
Premature physial closure; variable short stature; short metacarpals, metatarsals, and phalanges; and variable shortening of humerus, radius, and ulna. Hypersegmentation with an extra ossicle producing phalangeal deviation distinguishes type C.
BRACHYOLMIA (BRACHYRHACHIA)
Named from Greek βραχvζ: “short” and ολµοζ: “trunk,” whence the synonym brachyrhachia, from Greek ραχιζ: “spine”.
Affects the spine less, and is associated with precocious calcification of the falx cerebri.
3
Autosomal dominant, caused by a gain of function mutation in the gene for transient receptor potential cation channel subfamily V member 4, a Ca2+ channel. Allelic with Charcot-Marie-Tooth and spinal muscular atrophy, distal subtype.
Kyphoscoliosis and flattened, irregular cervical vertebrae.
4
Autosomal recessive, caused by mutation in gene encoding enzyme bifunctional 3′-phosphoadenosine 5′-phosphosulfate synthetase 2. The enzyme synthesizes 3′-phosphoadenosine 5′-phosphosulfate from ATP and inorganic sulfate, providing the source for cellular sulfation.
BRUCK SYNDROME
Type
Mutation
1
Mutation of FKBP10 gene on 17q21.
2
Mutation of PLOD2 gene on 3q23-q24.
Deficiency of bone-specific telopeptide lysyl hydroxylase, resulting in aberrant cross-linking of type I collagen. Lysine residues in the triple helix are normally hydroxylated. Enzyme normal in cartilage and ligament.
Fractures and Wormian bones resemble osteogenesis imperfecta. Normal sclerae and teeth. Contractures resemble arthrogryposis, hence the appellation “osteogenesis imperfecta with joint contractures.” Pterygia, scoliosis, and clubfoot.
CAFFEY DISEASE (INFANTILE CORTICAL HYPEROSTOSIS)
Mutation of 17q21.31-q22, which encodes the α-1 chain of type 1 collagen; however, no features of osteogenesis imperfecta.
Autosomal dominant as well as de novo mutation.
Onset in the first few months of life with spontaneous resolution by 2 years with minimal sequelae. Despite its name, it has been detected by ultrasonogram in utero (prenatal form) and in adulthood.
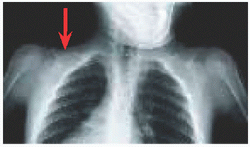
Inflammatory presentation, including fever and hot, tender long bones [A] and mandibles [B], which show diaphysial periosteal deposition.
This is distinct among hereditary disorders in being transient and leaving no residue.
CAMP(T)OMELIC DYSPLASIA
Greek καµπη: “bending, flexion, and twisting” and µελοσ: “limb,” to describe the characteristic feature of long bone bowing, especially of the tibiae, clubfoot, and hip dislocation.
17q24 mutation with haploinsufficiency of SOX 9.
Cutaneous dimpling at apex of bowing. Cleft palate, micrognathia, flat face, and pterygium colli. Thoracic dysplasia, including tracheobronchial hypoplasia, bladeless scapulae, slender or absent ribs, reduced cage volume, and sternal mineralization. Congenital heart and kidney disease. Death is frequent in infancy due to respiratory insufficiency.
Gonadal dysgenesis may culminate in sex reversal of affected XY cases.
CARPENTER SYNDROME
Autosomal recessive mutation in Ras-associated protein RAB23 gene on 6p11.
Also called acrocephalopolysyndactyly. Craniosynostosis produces a “pointed head.” Brachysyndactyly of the hands and preaxial polysyndactyly of the feet. Correction of genu valgum to stabilize patellae. Pilonidal dimple with absent coccyx.
Variable mental retardation, short stature, obesity, and eye, ear, cardiovascular, and genitourinary anomalies.
CHONDRODYSPLASIA PUNCTATA (CONRADIHÜNERMANN)
Dominant mutation in the gene encoding delta(8)-delta(7) sterol isomerase emopamil-binding protein on Xp11.23-p11.22, an enzyme essential to cholesterol biosynthesis. Rhizomicromelic dwarfism characterized by asymmetry of involvement and by calcific stippling of trachea, thorax, spine, pelvis, coracoid process, and glenoidal cavity. The latter typically resolves after first year of life. Kyphoscoliosis and clubfoot. Cutaneous disease, including striated ichthyosiform hyperkeratosis, whorled pigmentation, cicatricial alopecia, and “orange-peel” skin. Ocular anomalies, including cataracts, nystagmus, and glaucoma. Warfarin teratogenicity, by inhibition of synthesis of gamma-carboxyglutamic acid, which is involved in both clotting and calcification, may lead to chondrodysplasia punctata.
CHONDROECTODERMAL DYSPLASIA (ELLIS-VAN CREVELD)
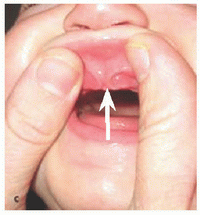
Mutation in Ellis-van Creveld gene on 4p16. Micromelic dwarfism characterized by postaxial polydactyly, capitate-hamate fusion, genua valga, clubfoot, nail dystrophy, and rib hypoplasia with narrow chest. Upper lip anomaly described as “lip-tie” [A] and tooth eruption at birth described as “natal teeth.” Cardiac and male genitourinary anomalies and variable mental retardation. Largest pedigree in the Old Order Amish of Lancaster County, Pennsylvania, whose members were described as having “six-fingered dwarfism.” On the way to a pædiatric conference in England (1938), the Scott Ellis met the Dutchman van Creveld while sitting in the same compartment of a train, where they discussed a case they each had seen independently.
CLEIDOCRANIAL DYSPLASIA
Autosomal dominant loss-of-function mutation in runt-related transcription factor 2 gene (RUNX2) on 6p21.1.
Head has been likened to a “hot cross bun” due to persistent open sutures. The head also is known as “Arnold” head, after a Muslim Chinese progenitor from South Africa with >1,000 descendants.
Midline defects, including hypoplastic or aplastic clavicles with hypermobile shoulders, short middle phalanges, coxa vara, symphysis pubis diastasis, scoliosis, and spondylolisthesis. Dental anomalies.
Formerly called “dysostosis” to emphasize the regional nature of anomalies of the head and shoulder.
CORNELIA DE LANGE SYNDROME
Autosomal dominant as well as de novo mutation in Nipped B-like (NIPBL) gene on 5p13.1, which encodes a component of cohesin, a protein complex that coheres sister chromatids during cell division. Characteristic facies, including synophrys, crescentic, or “carp” mouth, long philtrum, anteverted nares, and ptosis.
Mental and growth retardation, “growling” cry, hirsutism, and ocular, cardiac, genitourinary, gastrointestinal, and pulmonary anomalies. Self-injurious and autistic behavior. Micromelia disproportionately affecting the upper limb, including ulnar dysgenesis, radial head dislocation, oligodactyly, proximally placed thumb, clinodactyly of smallest finger, and single palmar flexion crease.
de Lange was Professor of Pædiatrics at the University of Amsterdam, where she was followed by van Creveld. The disorder was described 17 years earlier (1916) by Brachmann, whose studies were interrupted by a call to the German Army.
CRANIODIAPHYSIAL DYSPLASIA
Mutation in the SOST gene on 17q12-q21.
Hyperostosis of skull encroaches on foramina and osseous canals leading to cranial nerve palsy and hearing loss, of face results in “leonine facies,” and of the skeleton bones produces diaphysial sclerosis and medullary stenosis, in particular of ribs, clavicles, and sternum.
CRANIOMETAPHYSIAL DYSPLASIA
Autosomal dominant form caused by mutation in the human homolog of mouse progressive ankylosis gene on 5p15.2-p14.1. Additional autosomal recessive form mapped to chromosome 6q21-22.
Skull and facial manifestations similar though less severe than above. Metaphysial rather than diaphysial involvement is distinguished by “Erlenmeyer flask” deformity in long bones.
de BARSY SYNDROME
This is one of the progeroid syndromes, which are distinguished by cutis laxa with subcutaneous paucity of fat and prominence of veins, together with “pseudohydrocephalic” head, producing an “old appearance,” from Greek γερωv: “old man.”
This type most affects the skeleton: multiple joint dislocations and subluxations, in particular of the hip, scoliosis, and vertical talus.
Other features include corneal clouding, short stature, and mental retardation.
DIASTROPHIC DYSPLASIA
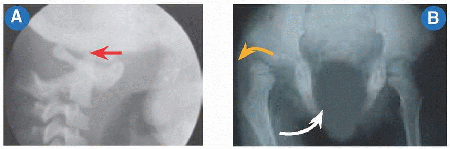
Mutation in the solute carrier family 26 (sulfate transporter), member 2, gene (SLC26A2) on 5q32-q33.1. Allelic to epiphysial dysplasia, multiple. Greek δια-, an emphatic prefix, and στρεφω, “I twist,” describe the “severely twisted” clubfeet and spine. The latter includes thoracolumbar kyphoscoliosis and cervical kyphosis [A]. Short first metacarpal producing “hitchhiker thumb” [B] and calcification of pinnae producing “cauliflower ear.” The former permits diagnosis on ultrasonogram at 16 months in utero. Multiple joint contractures and malformations, in particular of the hips, which show flattening and a “double-hump” deformation.
Other features include cleft palate, collapse of the tracheobronchial tree, and restrictive pulmonary disease.
DOWN SYNDROME (TRISOMY 21)
Genomic dosage imbalance at 21q22.3 producing phenotypic variability. Diagnosis by Quad test (serum α-fetoprotein, estriol, β-HCG, and inhibin A) in second trimester of pregnancy: a positive test is followed by amniocentesis.
Risk increases with maternal age: 9-fold from 30 years to 40 years. Incidence is 1:1,000.
“Mongoloid” facies; simian (Latin simia: “ape”) crease (single transverse palmar); hypotonia; ligamentous laxity; instability of C1-C2 [A], hips [B], and patella; flat feet; and scoliosis.
Screen for C1-C2 instability in symptomatic patient, including local signs, such as torticollis and neck stiffness, and global signs, such as myelopathy. Asymptomatic or general screening is contraindicated:
Symptoms and signs precede neural injury.
Radiographic instability may alternate with stability without clinical correlation.
Atlantoaxial fusion has a high complication rate.
Cognitive impairment; hearing loss, usually conductive; hypothyroidism; and congenital malformation of heart, in particular atrioventricular septal defect; gut, such as duodenal atresia; blood, in particular leukemia; and brain, including senile plaques and neurofibrillary tangles leading to premature Alzheimer disease.
The disorder was first described by John Langdon Haydon Down of London, whose Observations on an Ethnic Classification of Idiots (1866) included a description of the “Mongoloid” type. While this study has been condemned as racist, the final sentence suggests no such intention, as Down regarded the “degeneracy” across racial barriers “to furnish some arguments in favour of the unity of the human species.”
Down also first described a mentally delayed obese girl whose hands and feet remained small as a hypogonadal adult, seven decades before the report of Prader, Labhart, and Willi.
DYGGVE-MELCHIOR-CLAUSEN SYNDROME
Mutation in dymeclin gene at 18q21.1.
Dymeclin is necessary for correct organization of Golgi apparatus. Allelic with Smith-McCort dysplasia. Psychomotor retardation, hip instability with waddling gait, odontoid hypoplasia with C1-C2 instability, platyspondyly, vertebral anterior beaking and kyphoscoliosis, hypoplasia of scapula and glenoid cavity, epiphysial/apophysial irregularity manifesting as “lace-border” iliac crests, widening of sacroiliac joints and symphysis pubis, and camptodactyly.
DYSPLASIA EPIPHYSIALIS HEMIMELICA
Also known as Trevor disease. Non-Mendelian and nonfamilial. Boys more than thrice girls. Osteocartilaginous tumors arising from epiphysis, often lower limb, multilevel, and ipsilateral. Lesions cause pain, swelling, and deformity, starting during infancy or early childhood. Radiolucency of lesions delays diagnosis. Manage by excision and osteotomy for deformity correction. Recurrence is common, necessitating repeat operation(s).
EHLERS-DANLOS SYNDROMES
A group of hereditable disorders characterized by:
Skin hyperextensibility. Acrogeria. Collagen fibers seen on electron microscopy of skin have been likened to hieroglyphics.
Articular hypermobility. Instability of hip, patella, elbow, and shoulder.
Tissue fragility. Prematurity in 50% due to premature rupture of fetal membranes. Vascular and visceral rupture. Bruisable skin that heals with “cigarette paper” scars. Hernia and pneumothorax.
Skeletal deformity. Kyphoscoliosis, spondylolisthesis, atlantoaxial rotatory displacement, flatfoot, and pectus deformity. Tendency to recurrent deformity after correction.
The disorder may be classified into 10 subtypes, but V, VIII, and X may not be distinct entities. The subtypes have distinguishing features, with variable overlap. Most have autosomal dominant transmission.
Type
Mutation
I
Type V collagen α-1 chain on 9q34.3.
Type V collagen α-2 chain on 2q32.2.
Type I collagen α-1 chain on 17q21.33.
II
Type V collagen α-1 chain on 9q34.3.
III
Tenascin-XB on 6p21.3.
Type III collagen α-1 chain on 2q32.2.
IV
Type III collagen α-1 chain on 2q32.2.
V
Abnormal collagen cross-linking due to deficiency of lysyl hydroxylase.
VI
Lysyl hydroxylase on 1p36.22.
VII
Type I procollagen N-proteinase on 5q35.3.
VIII
12p13.
IX
Cu(2+)-transporting ATPase, alpha polypeptide on Xq21.1. Allelic to Menkes syndrome.
X
Fibronectin.
Type
Features
I
Classic gravis: “severe”.
II
Classic mitis: “mild”. Mildness may delay or preclude diagnosis.
III
Hypermobility without skeletal deformity.
IV
Vascular. Autosomal dominant or recessive. Spontaneous rupture of major vessels and viscera. Aneurysm, fistula.
V
X-linked.
VI
Ocular-scoliotic. Kyphoscoliosis from infancy. Retinal detachment, scleral fragility, rupture of ocular globe.
VII
Dermatosparaxis (“skin tearing”) due to abnormal type I collagen in skin. Autosomal recessive.
VIII
Peri-odontitis: gingival recession, premature loss of teeth, resorption of alveolar bone.
Striae distensae. Petechiae due to defect in platelet aggregation.
EMANUEL SYNDROME
Malsegregation of the t(11;22)(q23;q11.2) translocation, a rare example in humans of reciprocal (non-Robertsonian) exchange of genetic material between chromosomes.
Kyphosis and scoliosis and hip dislocation.
Ear: preauricular tag and sinus, low set, hearing loss, and otitis media.
Eyes: hooded eyelids, strabismus, and myopia.
Psychomotor delay; seizures; cardiovascular anomalies, including aortic and pulmonary stenosis and septal defects; and genitourinary anomalies, including absent kidney and cryptorchidism.
EPIPHYSIAL DYSPLASIA, MULTIPLE
Genetic heterogeneity manifested by six types, designated EDM1-6.
Type
Features
1
Mutation in the gene for cartilage oligomeric matrix protein on 19p13.11. Includes milder form (Ribbing) and more severe form (Fairbank). Allelic to pseudoachondroplasia.
Diagnosis aided by reduced serum levels of cartilage oligomeric matrix protein
2
Mutation in gene encoding type 9 collagen α-2 chain on 1p33-p32.2, which also has been implicated in susceptibility to intervertebral disc disease with sciatica.
3
Mutation in gene encoding type 9 collagen α-3 chain on 20q13.33. Myopathy may distinguish this type.
4
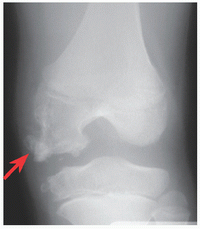
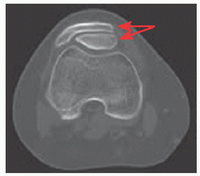
Mutation in the solute carrier family 26 (sulfate transporter), member 2 gene (SLC26A2) on 5q32-q33.1. Allelic to diastrophic dysplasia, atelosteogenesis II, achondrogenesis IB. Distinguished by clubfoot and double-layered patella (red arrows).
5
Mutation in matrilin-3 gene on 2p24.1.
Allelic to one form of spondylo-epimetaphysial dysplasia.
6
Mutation in gene encoding type 9 collagen α-1 chain on 6q13.
Normal to moderate short stature. Delayed and irregular epiphysial formation leads to long bone deformity, in particular coxa vara, genua vara or valga, and brachydactyly as well as premature osteoarthritis.
Multiple epiphysial involvement distinguishes this disorder from Legg-Calvé-Perthes disease. Sparing of the spine distinguishes it from spondyloepiphysial dysplasia.
ESCOBAR SYNDROME
See pterygium syndrome.
FAMILIAL DYSAUTONOMIA
Also known as Riley-Day syndrome, congenital insensitivity to pain, and hereditary sensory and autonomic neuropathy type III.
Mutations in the inhibitor of kappa light polypeptide gene enhancer in B cells, kinase complex-associated protein (IKBKAP) gene on 9q31.3.
Diminished pain and temperature perception leads to self-inflicted injuries. Vasomotor instability often triggered by stress, hyperhidrosis, alacrima, cutaneous blotching, and absence of fungiform papillae on tongue. Lack of axon flare after intradermal injection of histamine. Gastrointestinal and renal dysfunction. Increased prevalence in Ashkenazi Jewish descent.
Orthopedic problems include fracture, autoamputation, osteomyelitis, septic arthritis, neuropathic arthropathy, vibratory loss, areflexia, and scoliosis.
Emotional lability and absence of pain dictate conservative management.
FANCONI ANÆMIA
Genetically heterogeneous with 15 complementation groups. Common feature is abnormal DNA breakage, cross-linking, and repair.
Myelophthisis with pancytopenia requires bone marrow transplant.
Short stature. Radial defects, including hypoplastic/absent/bifid thumb as well as absent radius, require reconstruction.
Genitourinary anomalies, including hypoplastic/absent/horseshoe/ectopic kidney, hypogonadism, cardiac septal defects, hyperpigmentation with café au lait spots, and malignant diathesis.
FEMORAL-FACIAL SYNDROME
Facies characterized by long philtrum, thin upper lip, hypoplastic alae nasi, and microretrognathia.
Femoral hypoplasia/aplasia with acetabular dysplasia. Radioulnar and radiohumeral synostosis. Congenital scoliosis. Sacral dysplasia may resemble caudal regression syndrome. Feet with preaxial polysyndactyly and clubfoot. Sprengel anomaly.
Cardiac and genitourinary anomalies.
One-third of patients have a prenatal history of maternal diabetes.
FIBRODYSPLASIA OSSIFICANS PROGRESSIVA
Distinguish myositis ossificans, a general term for heterotopic ossification that may be subclassified as traumatica when it follows injury and is not hereditable.
Mutation in activin A receptor type I gene on 2q24.1 results in abnormal signal transduction in response to bone morphogenetic protein type I.
Episodic and unpredictable heterotopic ossification of striated muscle, in craniocaudad, axial to appendicular, and proximal to distal directions.
Only signs at birth are halluceal deformation and monophalangism. Clinodactyly, digital reduction defects, vertebral fusion, hearing loss, and alopecia.
Pain and ankylosis, which may be exacerbated by trauma (both accidental and iatrogenic).
Mean age of onset 5 years; confinement to wheelchair by third decade.
Restrictive pulmonary disease may lead to respiratory failure.
Eighty percent of patients receive an incorrect initial diagnosis. Diagnosis is clinical: while lesions may be confused with malignancy, avoid biopsy as it exacerbates condition.
FREEMAN-SHELDON SYNDROME
Also known as whistling face-windmill vane hand syndrome and craniocarpotarsal dystrophy.
Mutation in embryonic skeletal muscle myosin heavy chain 3 gene on 17p13.1.
A type of distal arthrogryposis.
Small mouth with pursed lips resembles whistling. Camptodactyly with ulnar deviation has been likened to a windmill vanes. Kyphoscoliosis. Contractures of hips (without or with dislocation), knees, and shoulders. Clubfoot and vertical talus.
Myopathy and seizure. Malignant hyperthermia may impact operation.
FRIEDREICH ATAXIA
Mutation in frataxin gene on 9q21.11. Second locus on 9p reflects genetic heterogeneity. Frataxin is involved in mitochondrial iron homeostasis.
Autosomal recessive. Most common inherited ataxia.
Ataxia, absent deep tendon reflexes, impaired proprioception and vibratory sense, dysarthria, extensor plantar response (Babinski), and nystagmus.
Pes cavus and scoliosis.
Preadolescent onset; confinement to wheelchair by fourth decade.
Hypertrophic cardiomyopathy: death most frequently from heart failure.
GAUCHER DISEASE
Mutation in gene encoding acid β-glucosidase on 1q22.
Cells of mononuclear phagocyte origin (such as macrophages) laden with glucosylceramide (glucosylcerebroside), known as Gaucher cells, accumulate in bone marrow, spleen, liver, lung, ocular limbus, and skin, leading to pancytopenia, hepatosplenomegaly, interstitial restrictive lung disease, pingueculae, and cutaneous pigmentation.
Continuum and wide spectrum of severity: perinatal lethal to asymptomatic adult.
Osteolysis, bone crises, and pathologic fractures. Widening of distal metaphysis of femur likened to “Erlenmeyer flask” (orange). Osteonecrosis of head of femur managed by hip arthroplasty.
Type
Features
1
Non-neuropathic.
2
Neuropathic—acute.
Perinatal or infantile lethal.
3
Neuropathic—chronic.
Later onset, slower progression.
Partial splenectomy for thrombocytopenia, to balance risk of sepsis.
Multifaceted treatment includes enzyme replacement, chemical chaperone, substrate reduction, and bone marrow transplantation.
Increased prevalence in Ashkenazi Jewish descent.
GOLDENHAR SYNDROME
Linked to 14q32.
Also known as oculoauriculovertebral dysplasia and hemifacial microsomia.
Anomalies of first and second branchial arch derivatives.
Facial reconstruction for asymmetric eye and ear anomalies.
Spine fusion for congenital scoliosis. Reconstruction for radial ray anomalies, which are ipsilateral to facial anomalies.
Congenital heart disease, including tetralogy of Fallot and coarctation of aorta. Central nervous system lesions, including hydrocephalus and cerebellar hypoplasia. Genitourinary anomalies, including multicystic or ectopic kidney.
GUILLAIN-BARRÉ SYNDROME
Familial type caused by mutation in the peripheral myelin protein 22 gene on 17p12. Allelic with Charcot-Marie-Tooth disease type 1.
Acute demyelinating polyneuropathy resulting from aberrant immune mechanism suggested by preceding upper respiratory infection or Campylobacter jejuni enteritis.
Ascending symmetric flaccid paralysis, proximal muscles more affected, ophthalmoplegia, and dysphagia. Variable sensory involvement, including loss of proprioception, and autonomic dysfunction, such as arrhythmia.
Involvement of respiratory muscles may necessitate ventilator support.
Cerebrospinal fluid analysis shows albuminocytologic dissociation: elevated protein without elevated cell count, in contrast with infection.
Treat with plasmapheresis or immunoglobulin per venam.
HAND-FOOT-GENITAL SYNDROME
Mutation in the homeobox A13 gene on 7p15.2.
Genitourinary anomalies, including double uterus and bifid scrotum.
Short first metacarpal and metatarsal result in proximal location of hypoplastic thumbs and halluces. Smallest finger clinodactyly-brachydactyly. Carpal and tarsal fusions.
HÆMOPHILIA
Type
Features
A
Mutation in gene encoding coagulation factor VIII on Xq28.
Recessive affects boys.
Mild (40% of cases): 6-30% factor level, hæmorrhage after trauma.
Moderate (10%): 1-5%.
Severe (50%): <1%, at least monthly spontaneous hæmorrhage.
1:10000
B Christmas disease
Mutation in gene encoding coagulation factor XI on Xq27.1.
Recessive.
1:30,000
Named after patient Stephen Christmas (1947-1993). B(M): inhibition of factor VII by abnormal factor IX prolongs PT.
B Leyden: factor IX increases after puberty to eliminate hæmorrhagic diathesis.
Laboratory tests show normal platelet count and prothrombin time (PT), but a prolonged activated partial thromboplastin time (aPTT).
Hemorrhage into joints and muscles, in contrast with bleeding disorders due to platelet defects or von Willebrand disease, in which mucosal bleeding predominates.
Hemarthrosis begins after walking and is characterized by swelling, pain, stiffness, and inflammatory arthritis. Muscle hemorrhage causes necrosis, contractures, and neuropathy by entrapment.
Only gold members can continue reading. Log In or Register to continue