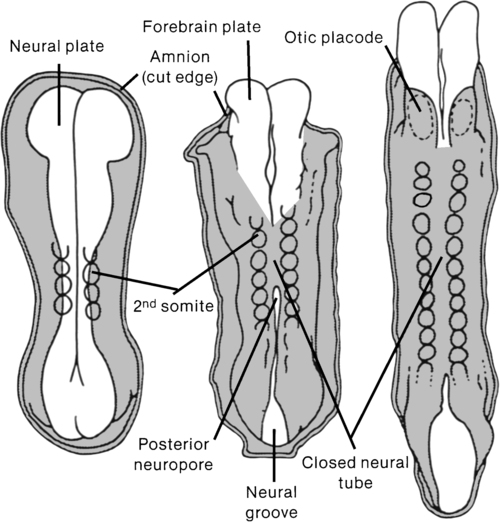
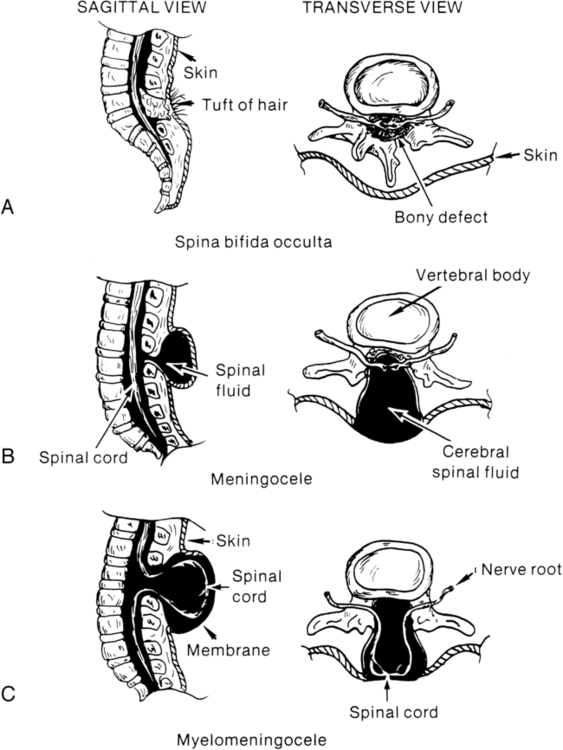
KRISTIN J. KROSSCHELL, PT, MA, PCS and MARI JO PESAVENTO, PT, PCS After reading this chapter the student or therapist will be able to: 1. Identify the various types of spina bifida. 2. Recognize the incidence and etiology of spina bifida. 3. Identify the clinical manifestations of myelomeningocele, including neurological, orthopedic, and urological sequelae. 4. Comprehend medical management in the newborn period and beyond. 5. Determine physical and occupational therapy evaluations, including manual muscle testing, range of motion, sensory testing, reflex testing, developmental and functional and mobility assessments, and perceptual and cognitive evaluations. 6. List the major physical and occupational therapy goals and appropriate therapeutic management for each of the following stages: (a) before surgical closure of sac, (b) after surgery during hospitalization, (c) preambulatory, (d) toddler through preschool age, (e) primary school age through adolescence, and (f) transition to adulthood. 7. Identify psychological adjustment to congenital spinal cord injury. A developmental framework, the Guide to Physical Therapist Practice,1 and the International Classification of Functioning, Disability and Health (ICF) have been used to aid in understanding the sequential problems of the child with spina bifida. The developmental model, however, must always stay in line with the functional model for adult trauma because the problems of the congenitally involved child grow quickly into limitations in functional activities and participation in life of the injured adult. With concentration on the present but with an eye to the future, appropriate management goals can be achieved. A congenital spinal cord lesion occurs in utero and is present at the time of birth. Understanding how this malformation develops requires an appreciation of normal nervous system maturation. The nervous system develops from a portion of embryonic ectoderm called the neural plate. During gestation, the neural plate develops folds that begin to close, forming the neural tube (Figure 15-1). The neural tube differentiates into the CNS, which is composed of brain and spinal cord tissue. In the normal embryo, neural tube closure begins in the cervical region and proceeds cranially and caudally. Closure is generally complete by the twenty-sixth day. Spina bifida involves a defect in the neural tube closure and the overlying posterior vertebral arches. The extent of the defect may result in one of two types of spina bifida: occulta or cystica. Spina bifida occulta is characterized by a failure of one or more of the vertebral arches to meet and fuse in the third month of development. The spinal cord and meninges are unharmed and remain within the vertebral canal (Figure 15-2, A). The bony defect is covered with skin that may be marked by a dimple, pigmentation, or patch of hair.2 The common site for this defect is the lumbosacral area, and it is usually associated with no disturbance of neurological or musculoskeletal functioning. Spina bifida cystica results when the neural and overlying vertebral arches fail to close appropriately. Cystic protrusion of the meninges or the spinal cord and meninges is present through the defective vertebral arches. The milder form of spina bifida cystica, called meningocele, involves protrusion of the meninges and cerebrospinal fluid (CSF) only into the cystic sac (see Figure 15-2, B). The spinal cord remains within the vertebral canal, but it may exhibit abnormalities.3 Clinical signs vary (according to spinal cord anomalies) or may not be apparent. This is a relatively uncommon form of spina bifida cystica. A more severe form of spina bifida cystica, called myelocele or myelocystocele, is present when the central canal of the spinal cord is dilated, producing a large, skin-covered cyst. The neural tube appears to close normally but is distended from the cystic swelling. The CSF may ceaselessly expand the neural canal. Prompt medical attention is mandatory. This form of spina bifida is also rare.4 The more common and severe form of the defect is known as myelomeningocele, in which both spinal cord and meninges are contained in the cystic sac (see Figure 15-2, C). Within the sac the spinal cord and associated neural tissue show extensive abnormalities. In incomplete closure of the neural tube (dysraphism), abnormal growth of the cord and a tortuous pathway of neural elements make normal transmission of nervous impulses abnormal. The result is a variable sensory and motor impairment at the level of the lesion and below.2 In an open myelomeningocele, nerve roots and spinal cord may be exposed, with dura and skin evident at the margin of the lesion. Exposure of the open neural tube to the amniotic fluid environment leads to neuroepithelial degeneration, with massive loss of neural tissue by the end of pregnancy.5 Although spina bifida cystica can occur at any level of the spinal cord, myelomeningoceles are most common in the thoracic and lumbosacral regions. Myelomeningocele occurs in 94% of the cases of spina bifida cystica, and two thirds of open lesions involve the thoracolumbar junction.2 The terms spina bifida, myelodysplasia, and myelomeningocele are frequently used interchangeably. Other forms of spinal dysraphism include diastematomyelia, lipomeningocele, and sacral agenesis. Diastematomyelia is present in 30% to 40% of patients with myelomeningocele and is secondary to partial or complete clefting of the spinal cord.6 Lipomeningocele, another form of spina bifida cystica, is usually caused by a vertebral defect associated with a superficial fatty mass (lipoma or fatty tumor) that merges with the lower level of the spinal cord. No associated hydrocephalus is present, and neurological deficit is generally minimal; however, problems with urinary control and motor control of the lower extremities may be noted.7 Neurological tissue invasion may be caused by a tethered spinal cord; therefore early lipoma resection is indicated for cosmesis and to minimize neurological sequelae. Lumbosacral or sacral agenesis may occur and is caused by an absence of the caudal part of the spine and sacrum. Children with this form of dysraphism may have narrow, flattened buttocks, weak gluteal muscles, and a shortened intergluteal cleft. The normal lumbar lordosis is absent, although the lower lumbar spine may be prominent. Calf muscles may be atrophic or absent. The pelvic ring is completed with either direct opposition of the iliac bones or with interposition of the lumbar spine replacing the absent sacrum. These children may have scoliosis, motor and sensory loss, and visceral abnormalities including anal atresia, fused kidneys, and congenital heart malformations. Management is started early and is symptomatic for each system.8 Failure of fusion of the cranial end of the neural tube results in a condition known as anencephaly. In this condition some brain tissue may be evident, but forebrain development is usually absent.9 Sustained life is not possible with this neural tube defect; therefore this condition is not discussed further. Statistics about the incidence of spina bifida vary considerably in different parts of the world. Spina bifida and anencephaly, the most common forms of neural tube defects, affect about 300,000 newborns each year worldwide.10 In the United States the incidence is currently 2.48 per 10,000, down from approximately 7.23 per 10,000 births from 1974 through 1979 (before the folic acid mandate).11,12 Current worldwide folic acid fortification programs have resulted in decreased incidence of spina bifida,13,14 with annual decreases of 6600 folic acid–preventable spina bifida and anencephaly births reported since 2006.15 There was a 31% decline in spina bifida prevalence rates in the immediate postfortification period (October 1998 through December 1999).13 There was a continued decline in spina bifida prevalence rates from 1999 to 2004 of 10%.16 Studies have also demonstrated that decline varied by ethnicity and race from prefortification to optional fortification to mandatory fortification in the United States.16,17 Initially after fortification, the largest decline in prevalence was noted in Hispanic and non-Hispanic white races or ethnicities. Despite this initial decline, postfortification prevalence rates remain highest in infants born to Hispanic mothers, and less in infants born to non-Hispanic white and non-Hispanic black mothers.16 In addition to periconceptual folate supplementation, it is thought that incidence has decreased subsequent to food fortification in several countries, decreased exposure to environmental teratogens, and increased and more accurate prenatal screening for fetal anomalies.10 Spina bifida is thought to be more common in females than in males, although some studies suggest no real sex difference.3 A study of the association of race and sex with different neurological levels of myelomeningocele found the proportions of whites and females to be significantly higher in patients with thoracic-level spina bifida.4 A significant relation also has been noted between social class and spina bifida: the lower the social class, the higher the incidence.18,19 A multifactorial genetic inheritance has been proposed as the cause of spina bifida, coupled with environmental factors, of which nutrition, including folic acid intake, are key. Cytoplasmic factors, polygenic or oligogenic inheritance, chromosomal aberrations, and environmental influences (e.g., teratogens) have all been considered as possible causes.5,15 Genetic factors seem to influence the occurrence of spina bifida. The chances of having a second affected child are between 1% and 2%, whereas in the general population the percentage drops to one fifth of 1%.20,21 Although these factors are related to the incidence of spina bifida, the cause of this defect remains in question. Environmental conditions, such as hyperthermia in the first weeks of pregnancy, or dietary factors, such as eating canned meats or potatoes or drinking tea, have been implicated but not substantiated.22,23 In addition, historically, nutritional deficiencies, such as of folic acid and vitamin A, have been implicated as a cause of primary neural tube defects.24–27 Approximately 50% to 70% of neural tube defects can be prevented if a woman of childbearing age consumes sufficient folic acid daily before conception and throughout the first trimester of pregnancy. As a result of research findings in support of folic acid implementation, the U.S. Public Health Service has mandated folic acid fortification since 1998 as a public health strategy. Prenatal vitamins, especially folic acid, are recommended to discourage the condition’s development. Current fortification programs are preventing about 22,000 cases, or 9% of the estimated folic acid–preventable spina bifida and anencephaly cases.15 Genetic considerations, such as an Rh blood type, a specific gene type (HLA-B27), an X-linked gene, and variations in the many folate pathway genes have been implicated, but not conclusively.28,29 Malformations are attributed to abnormal interaction of several regulating and modifying genes in early fetal development.30 Disturbance of any of the sequential events of embryonic neurulation produces neural tube defects (NTDs), with the phenotype (i.e., spina bifida, anencephaly) varying depending on the region of the neural tube that remains exposed.5 Environmental factors combined with genetic predisposition appear to trigger the development of spina bifida, although definitive evidence is not available to support this claim.31 The incidence of spina bifida has declined since the advent of amniocentesis and the use of ultrasonography for prenatal screening. The presence of significant levels of alpha fetoprotein in the amniotic fluid has led to the detection of large numbers of affected fetuses.32 Currently, maternal serum alpha-fetoprotein levels have been effective in detecting approximately 80% of neural tube defects.33 Prenatal screening can be most effective when a combination of serum levels, amniocentesis or amniography, and ultrasonography is used.34–36 Although this screening is not yet performed routinely, it is suggested for those at risk for the defect. Knowledge of the defect allows for preparation for cesarean birth and immediate postnatal care. This includes mobilization of the interdisciplinary team that will continue to care for the child. For parents who decide to carry an involved fetus to term, adjustment to their child’s disability can begin before birth, which includes mobilizing their own support system. Education from an integrated team regarding what will follow after delivery and neurosurgical closure is imperative to aid families in decision making and to allow families to assess and understand the child’s disability and future care options. Other advances in the field of prenatal medicine that affect spina bifida management and outcome include in utero treatment of hydrocephalus and in utero surgical repair to close the myelomeningocele. This challenging surgical procedure is practiced in only a few specialty centers and so far has been shown to offer palliation of the defect at best.37 Treatment such as this, in conjunction with prenatal diagnosis, has been shown to have a positive impact on the incidence and severity of complications associated with spina bifida.38–45 Limitations of current postnatal treatment strategies and considerations of prenatal treatment options continue to be explored. Ethics, timing of repair, and surgical procedures are all being investigated. In addition, continued assessment of outcomes from those who have undergone presurgical management requires continued exploration. The Management of Myelomeningocele Study (MOMS) was initiated in 2003 as a large randomized, clinical trial designed to compare the two approaches to the treatment of infants with spina bifida (prenatal or fetal surgery versus postnatal surgery) to determine if one approach was better than the other. The primary end point of this trial was the need for a shunt at one year, and secondary end points included neurologic function, cognitive outcome, and maternal morbidity after prenatal repair. This study had 112 patients enrolled in 2007 with a projected enrollment of 200.46–49 The trial was stopped for efficacy of prenatal surgery after enrollment of just 183 infants. Results demonstrated that prenatal surgery significantly reduced the need for shunting and improved mental and motor function at 30 months. Reduced incidence of hindbrain herniation at 12 months and successful ambulation by 30 months were also reported. While prenatal surgery was associated with improved function and reduced need for shunting, maternal and fetal risks, including preterm delivery and uterine dehiscense at delivery were reported.49a In 1996 the lifetime cost to society per affected person with spina bifida was estimated to be $635,000.50,51 More recent estimates have not been reported; however, with an economy in flux it is likely that this value underassesses costs to society today. In addition to medical management costs per child, there are additional costs that affect both the family and society across the life span that are variable and often related to differential market forces and social welfare policies.50 In 2007, Ouyang52 reported that average medical expenditures during the first year of life for those with spina bifida during 2002 and 2003 averaged $50,000 (using MarketScan 2003 database). The majority of expenditures during infancy were from inpatient admissions secondary to surgeries being concentrated during this time period for those with spina bifida. After infancy, average medical care expenditures during 2003 ranged from $15,000 to $16,000 per year among different age groups of persons with spina bifida. Incremental expenditures associated with medical care were not stable, but decreased with increasing age, from $14,000 per year for children to $10,000 per year for adults 45 to 64 years of age.52 The most obvious clinical manifestation of myelomeningocele is the loss of sensory and motor functions in the lower limbs. The extent of loss, while primarily dependent on the degree of the spinal cord abnormality, is secondarily dependent on a number of factors. These include the amount of traction or stretch resulting from the abnormally tethered spinal cord, the trauma to exposed neural tissue during delivery, and postnatal damage resulting from drying or infection of the neural plate.2 Specific clinical impairments that commonly lead to functional limitations for the child with spina bifida are addressed in this section. The orthopedic problems that occur with myelomeningocele may be the result of (1) the imbalance between muscle groups; (2) the effects of stress, posture, and gravity; and (3) associated congenital malformations. Decreased sensation and neurological complications also may lead to orthopedic abnormalities.53 Besides the obvious malformation of vertebrae at the site of the lesion, hemivertebrae and deformities of other vertebral bodies and their corresponding ribs also may be present.53,54 Lumbar kyphosis may be present as a result of the original deformity. In addition, as a result of the bifid vertebral bodies, the misaligned pull of the extensor muscles surrounding the deformity, as well as the unopposed flexor muscles, contributes further to the lumbar kyphosis. As the child grows, the weight of the trunk in the upright position also may be a contributing factor.54 Scoliosis may be present at birth because of vertebral abnormalities or may become evident as the child grows older. The incidence of scoliosis is lower in low lumbar or sacral level deformities.54,55 Scoliosis may also be neurogenic, secondary to weakness or asymmetrical spasticity of paraspinal muscles, tethered cord syndrome (TCS), or hydromyelia.55 Lordosis or lordoscoliosis is often found in the adolescent and is usually associated with hip flexion deformities and a large spinal defect.3,54 Many of these trunk and postural deformities exist at birth but are exacerbated by the effects of gravity as the child grows. They can compromise vital functions (cardiac and respiratory) and therefore should be closely monitored by the therapist and the family. As has been alluded to previously, the type and extent of deformity in the lower extremities depend on the muscles that are active or inactive. In total flaccid paralysis, in utero deformities may be present at birth, resulting from passive positioning within the womb. Equinovarus (clubfoot) and “rocker-bottom” deformity are two of the most common foot abnormalities. Knee flexion and extension contractures also may be present at birth. Other common deformities are hip flexion, adduction, and internal rotation, usually leading to a subluxed or dislocated hip. Although many of these problems may be present at birth, preventing positional deformity (such as the frog-leg position), which may result from improper positioning of flaccid extremities, is of the utmost importance. Orthopedic care varies throughout the course of the child’s life. Changes in clinical orthopedic management have evolved to establish evidence-based interventions.56 Because the paralyzed limbs of the child with spina bifida have increased amounts of unmineralized osteoid tissue, they are prone to fractures, particularly after periods of immobilization.57,58 Early mobilization and weight bearing can aid in decreasing osteoporosis.54,59 Fortunately, these fractures heal quickly with appropriate medical management. Hydrocephalus develops in 80% to 90% of children with myelomeningocele.21,60 Hydrocephalus results from a blockage of the normal flow of CSF between the ventricles and spinal canal. The most obvious effect of the buildup of CSF is abnormal increase in head size, which may be present at birth because of the great compliance of the cranial sutures in the fetus, or it may develop postnatally.61 Other signs of hydrocephalus include bulging fontanels and irritability. Internally, a concomitant dilation of the lateral ventricles and thinning of the cerebral white matter are usually present. Without reduction of the buildup of CSF, increased brain damage and death may result. Patients with myelomeningocele have a 99% chance of having an associated Chiari II malformation.6 Cardinal features of the Chiari II malformation include myelomeningocele in the thoracolumbar spine, venting of the intracranial CSF through the central canal, hypoplasia of the posterior fossa, herniation of the hindbrain into the cervical spinal canal, and compressive damage to cranial nerves. This malformation is a congenital anomaly of the hindbrain that involves herniation of the medulla and at times the pons, fourth ventricle, and inferior aspect of the cerebellum into the upper cervical canal. The herniation usually occurs between C1 and C4 but may extend down to T1.6,62,63 In those with Chiari II malformations and spina bifida there is a significant reduction in cerebellar volume, and within the cerebellum the anterior lobe is enlarged and the posterior lobe is reduced.64 Not all Chiari II malformations are symptomatic. As a result of a symptomatic Chiari malformation, problems with respiratory and bulbar function may be evident in the child with spina bifida.2 Paralysis of the vocal cords occurs in a small percentage of patients and is associated with respiratory stridor. Apneic episodes also may be evident, although their direct cause remains in question. Children with spina bifida also may exhibit difficulty in swallowing and have an abnormal gag reflex.2 Problems with aspiration, weakness and cry, and upper-extremity weakness also may be present in children with a symptomatic Chiari II malformation.65,66 Thus, depending on the orthopedic deformities present and the neurological involvement, severe respiratory involvement is possible in the affected child. These symptoms may be caused by significant compression of the hindbrain structures or dysplasia of posterior fossa contents, which can also occur in patients with Chiari II malformation.6,67 This complex hindbrain malformation is a common cause of death in children with myelomeningocele despite surgical intervention and aggressive medical management.68 Diffusion tensor tractography studies of association pathways in children with spina bifida have revealed characteristics of abnormal development, impairment in myelination, and abnormalities in intrinsic axonal characteristics and extraaxonal or extracellular space. These changes in diffusion metrics observed in children with spina bifida are suggestive of abnormal white matter development and persistent degeneration with increased age.69 Twenty percent to 80% of patients with myelomeningocele have hydromyelia.6,70,71 Hydromyelia signifies dilation of the center canal of the spinal cord as hydrocephalus signifies dilation of the ventricles of the brain. The area of hydromyelia may be focal, multiple, or diffuse, extending throughout the spinal cord. The hydromyelia may be a consequence of untreated or inadequately treated hydrocephalus with resultant transmission of CSF through the obex into the central canal, with distention a result of increased hydrostatic pressure from above.6 The increased collection of fluid may cause pressure necrosis of the spinal cord, leading to muscle weakness and scoliosis. Common symptoms of hydromyelia include rapidly progressive scoliosis, upper-extremity weakness, spasticity, and ascending motor loss in the lower extremities.6,72 Aggressive treatment of hydromyelia at the onset of clinical signs of increasing scoliosis is mandatory and may lead to improvement in or stabilization of the curve in 80% of cases. Surgical interventions may include revision of a CSF shunt, posterior cervical decompression, or a central canal to pleural cavity shunt with a flushing device.6,67 Tethered spinal cord is defined as a pathological fixation of the spinal cord in an abnormal caudal location (Figure 15-3). This fixation produces mechanical stretch, distortion, and ischemia with daily activities, growth, and development.73 Ischemic injury from traction of the conus directly correlates with degree of oxidative metabolism and degree of neurologic compromise. In addition to ischemic injury, traction of the conus by the filum may also mechanically alter the neuronal membranes, resulting in altered electrical activity.74–78 The presence of tethered cord syndrome (TCS) should be suspected in any patient with abnormal neurulation (including patients with myelomeningocele, lipomeningocele, dermal sinus, diastematomyelia, myelocystocele, tight filum terminale, and lumbosacral agenesis). Presenting symptoms may include decreased strength (often asymmetrical), development of lower-extremity spasticity, back pain at the site of sac closure, early development of or increasing degree of scoliosis (especially in the low lumbar or sacral level),79,80 or change in urological function.68,81–83 Approximately 10% to 30% of children will develop TCS after repair of a myelomeningocele. Because essentially all children with repaired myelomeningocele will have a tethered spinal cord, as demonstrated on magnetic resonance imaging (MRI), the diagnosis of TCS is made based on clinical criteria. The six common clinical presentations of TCS are increased weakness (55%), worsening gait (54%), scoliosis (51%), pain (32%), orthopedic deformity (11%), and urological dysfunction (6%).84 This clinical spectrum may be primarily associated with these dysraphic lesions or may be caused by spinal surgical procedures.73 The cord may be tethered by scar tissue or by an inclusion epidermoid or lipoma at the repair site.6 The primary goal of surgery is to detach the spinal cord where it is adherent to the thecal sac, relieving the stretch on the terminal portion of the cord. Surgery to untether the spinal cord (tethered cord release [TCR]) is performed to prevent further loss of muscle function, decrease the spasticity, help control the scoliosis,80,85 or relieve back pain.86,87 The effectiveness of a TCR may be demonstrated by an increase in muscle function, relief of back pain, and stabilization or reversal of scoliosis.80,85,87 It has been reported that scoliosis response to untethering and progression of scoliosis after untethering vary with location of tethering80,87 as well as Risser grade88 and Cobb angle.89 Those with Risser grade 3 to 5 and Cobb angle less than 40 degrees are less likely to experience curve progression after untethering. Those with Risser grades 0 to 2 and Cobb angle greater than 40 degrees are at higher risk of recurrence.74,89 Spasticity, however, is not always alleviated in all patients.90 Selective posterior rhizotomy has been advocated for patients whose persistent or progressive spastic status after tethered cord repair continues to interfere with their mobility and functional independence.68,70 Because of the usual involvement of the sacral plexus, the child with spina bifida commonly deals with some form of bowel and bladder dysfunction. Besides various forms of incontinence, incomplete emptying of the bladder remains a constant concern because infection of the urinary tract and possible kidney damage may result.91 Regulation of bowel evacuation must be established so that neither constipation nor diarrhea occurs. Negative social aspects of incontinence can be minimized by instituting intervention that emphasizes patient and family education and a regular, consistently timed, reflex-triggered bowel evacuation.92 The last major clinical manifestation resulting from the neurological involvement of myelomeningocele is impaired intellectual function. Although children with spina bifida without hydrocephalus may have normal intellectual potential, children with hydrocephalus, particularly those who have shunt infections, are likely to have below-average intelligence.93–95 These children often demonstrate learning disabilities and poor academic achievement.96 Even those with a normal IQ show moderate to severe visual-motor perceptual deficits.97 The inability to coordinate eye and hand movements affects learning and may interfere with activities of daily living (ADLs), such as buttoning a shirt or opening a lunchbox.98 Difficulties with spatial relations, body image, and development of hand dominance may also be evident.2,98 Children with myelomeningocele demonstrate poorer hand function than age-matched peers. This decreased hand function appears to be caused by cerebellar and cervical cord abnormalities rather than hydrocephalus or a cortical pathological condition (see Chapter 21).99 Prenatal studies have shown that the CNS as a whole is abnormally developed in fetuses with myelomeningocele.100–103 The impairment of intellectual and perceptual abilities has been linked to damage to the white matter caused by ventricular enlargement.2 This damage to association tracts, particularly in the frontal, occipital, and parietal areas, could account for the often severe perceptual-cognitive deficits noted in the child with spina bifida.69,104 Lesser involvement of the temporal areas may account for the preservation of speech, whereas the semantics of speech, which depends on association areas, is impaired. The “cocktail party speech” of children with spina bifida can be deceptive because they generally use well-constructed sentences and precocious vocabulary. A closer look, however, reveals a repetitive, inappropriate, and often meaningless use of language not associated with higher intellectual functioning. Research on learning difficulties in children with spina bifida and hydrocephalus suggests that many of these children experience difficulties. Tasks and skills affected include memory, reasoning, math, handwriting, organization, problem solving, attention, sensory integration, auditory processing, visual perception, and sequencing.101–103 Latex allergy and sensitivity have been noted with increasing frequency in children with myelomeningocele, with frequent reports of intraoperative anaphylaxis.105–109 These children have also been reported to have a higher than expected prevalence of atopic disease.110 A 1991 Food and Drug Administration Medical Bulletin estimated that 18% to 40% of patients with spina bifida demonstrate latex sensitivity,105,111 with others reporting an incidence of 20% to 67%.112,113 Within latex is 2% to 3% of a residual-free protein material that is thought to be the antigenic agent.107 Frequent exposure to this material results in the development of the immunoglobulin E antibody. Children with spina bifida are more likely to develop the immunoglobulin E sensitivity because of repeated parental or mucosal exposure to the latex antigen.114 Because of the risk of an anaphylactic reaction, exposure to any latex-containing products such as rubber gloves, therapy balls, pacifiers, spandex, dental dams, elastic or rubber bands, balloons, adhesive bandages, or exercise bands should be avoided. Latex-free gloves, therapy balls, treatment mats, and exercise bands are now widely available and should be considered for standard use in all clinics treating children with spina bifida. Spina bifida, even in the absence of multiple surgical interventions, may be an independent risk factor for latex sensitivity. Foods reported to be highly associated with latex allergy include avocado, banana, chestnut, and kiwi.115 Latex-free precautions from birth are more effective in preventing latex sensitization than are similar precautions instituted later in life.115–117 Latex sensitization decreased from 26.7% to 4.5% in children treated in a latex-free environment from birth.117 The presence of paralysis and lack of sensation on the skin places the child with spina bifida at major risk for pressure sores and decreased skin integrity. Various types of skin breakdown have occurred in 85% to 95% of all children with spina bifida by the time they reach young adulthood.118 Common areas at risk for pressure sores include the lower back, kyphotic or scoliotic prominences, heels, feet, toes, and perineum. A pressure sore may result from excessive skin pressure that can cause reduced capillary flow, tissue anoxia, and eventual skin necrosis. Excessive pressure may manifest itself early as reactive hyperemia, a blister, and later as an open sore or overt necrosis. Chronic, untreated sores may lead to osteomyelitis and eventual sepsis.110 Pressure sores often result in loss of time from school and work and can lead to financial hardship from medical treatment and hospitalizations. These negative consequences can largely be prevented with attention to education and instruction of the child and family. The goal of such education is to foster an understanding of the causes of skin breakdown and the necessary meticulous attention to skin care that must be carried out on a regular basis. Nutritional intake and weight gain and loss have been found to be problematic in children with myelomeningocele. Early on, infants with spina bifida may have feeding issues as a result of an impaired gag reflex, swallowing difficulties, and a high incidence of aspiration.2,66 Altered oral-motor function has been attributed to the Chiari II malformation.119 These impairments may lead to nutritional issues and delayed growth and weight gain. Speech, physical, and occupational therapists as a team are often needed to address these issues. Conversely, obesity can be a significant issue for children with spina bifida. This problem is complex and multifactorial.120 Mobility limitations and decreased energy expenditure result in lower physical activity levels. In addition, decreased lower limb mass diminishes the ability to burn calories, which leads to weight gain. Decreased caloric intake as well as a lifelong engagement in rewarding and physically challenging physical activities are both necessary to enhance weight control and control obesity. Children with myelomeningocele are short in stature. Growth in these children may be influenced by growth-retarding factors as a result of a neurological deficit such as tethered cord.121 Endocrine disorders and growth hormone deficiency have also been found to contribute to short stature in this population.122 As a result of complex CNS anomalies (midline defects, hydrocephalus, Arnold-Chiari malformation), these children are at risk for hypothalamopituitary dysfunction leading to growth hormone deficiency.123,124 Treatment with recombinant human growth hormone has proven successful in fostering growth acceleration in these children.123,125,126 Since the early 1960s the presence of a myelomeningocele has been treated as a life-threatening situation, and sac closure most often takes place within the first 24 to 48 hours of life.2,127 Recent advances in treatment have led to investigational treatment in utero to repair the defect before birth.38 The aim of either surgery is to replace the nervous tissue into the vertebral canal, cover the spinal defect, and achieve a watertight sac closure.128 This early management has decreased the possibility of infection and further injury to the exposed neural cord.24,128,129 Progressive hydrocephalus may be evident at birth in a small percentage of children born with myelomeningocele. A greater majority, however, have hydrocephalus 5 to 10 days after the back lesion has been closed.128,130–132 With the advent of computed tomography (CT), early diagnosis of hydrocephalus can be made in the newborn without the need for clinical examination. Although clinical signs are not always definitive, hydrocephalus may be suspected if (1) the fontanels become full, bulging, or tense; (2) the head circumference increases rapidly; (3) a separation of the coronal and sagittal sutures is palpable; (4) the infant’s eyes appear to look downward only, with the cornea prominent over the iris (“sunsetting sign”); and (5) the infant becomes irritable or lethargic and has a high-pitched cry, persistent vomiting, difficult feeding, or seizures (Table 15-1).21,61,133 TABLE 15-1 SIGNS AND SYMPTOMS OF SHUNT MALFUNCTION If the results of CT confirm hydrocephalus, a ventricular shunt is indicated. This procedure involves diverting the excess CSF from the ventricles to some site for absorption. In general, two types of procedures—the ventriculoatrial (VA) and ventriculoperitoneal (VP) shunt—are currently used, the latter being the most common (Figure 15-4). The shunt apparatus is constructed from Silastic tubing and consists of three parts: a proximal catheter, a distal catheter, and a one-way valve. As CSF is pumped from the ventricles toward its final destination, backflow is prevented by the valve system. In this manner intracranial pressure is controlled, CSF is regulated, and hydrocephalus is prevented from causing damage to brain structures. An alternate means of controlling hydrocephalus may be the use of endoscopic third ventriculostomy (EVT). EVT is a procedure that, in selected patients with obstructive hydrocephalus, allows egress of CSF from the ventricles to the subarachnoid space. This can decompress the ventricles and allow normal intracranial pressures and brain growth. This procedure is typically reserved for last resort.134 Unfortunately for children with spina bifida, their problems do not end after the back is surgically closed and a shunt is in place. Management strategies in the care of shunted hydrocephalus vary.135 Shunt complications occur frequently and require an average of two revisions before age 10 years.60 The most common causes of complications are shunt obstruction and infection.2,136 Revising the blocked end of the shunt can clear obstructions. Infections may be handled by external ventricular drainage and courses of antibiotic therapy followed by insertion of a new shunting system.2 The problem of separation of shunt components has been largely overcome by the use of a one-piece shunting system. The single-piece shunt decreases the complications of shunting procedures. Prophylactic antibiotic therapy 6 to 12 hours before surgery and 1 to 2 days postoperatively is effective in controlling infection for both sac repair and shunt insertion.71 This brief course of antibiotics has not led to resistant organisms. The main cause of death in children with myelomeningocele remains increased intracranial pressure and infections of the CNS. With the use of antibiotics, shunting, and early sac closure, the survival rate has increased from 20% to 85%.61,94,137 Initial newborn workup should include a urological assessment. The urology team aims to preserve renal function and promote efficient bladder management. An early start to therapy helps to preserve renal function for children with spina bifida.138 Initially, a renal and bladder ultrasound is performed to assess those structures.100 Urodynamic testing can be performed to determine any blockage in the lower urinary tract. Functioning of the bladder outlet and sphincters, as well as ureteric reflux, also can be evaluated. These tests, plus clinical observations of voiding patterns, help the urologist classify the infant’s bladder function. If the bladder has neither sensory nor motor supply, a constant flow of urine is present. In this case infection is rare because the bladder does not store urine and the sphincters are always open.139 If no sensation but some involuntary muscle control of the sphincter exists, the bladder will fill, but emptying will not occur properly. Overflow or stress incontinence results in dribbling urine until the pressure is relieved. Because of constant residual urine, infection is a potential problem and kidney damage may result.139 When some voluntary muscle control but no sensation is present, the bladder will fill and empty automatically. The child can eventually be taught to empty the bladder at regular intervals to avoid unnecessary accidents. A program of clean intermittent catheterization (CIC) done every 3 to 4 hours prevents infection and maintains the urological system.140–143 Parents are taught this method and can then begin to take on this aspect of their child’s care. At the age of 4 or 5 years, children with spina bifida can be taught CIC; thus they become independent in bladder care at a young age. Achieving this form of independence adds to the normal psychological development of these children. Some children may require urinary diversion through the abdominal wall (ileal conduit) or through the appendix (Mitrofanoff principle appendicovesicotomy)144–146 or other, less common methods, such as intravesical transurethral bladder stimulation, to handle their urinary condition.140,147 Although CIC is not possible for all children with spina bifida, it remains the method of choice for bladder management. Bowel management and training programs should be started early. Medications, enemas, and attention to fiber content in the diet are all of value in establishing a bowel management program. The Malone antegrade continence enema (ACE) procedure is an important adjunct in the case of adults and children with problems of fecal elimination in whom standard medical therapies have failed.148,149
Spina bifida: A congenital spinal cord injury
Overview of congenital spinal cord injury
Types of spina bifida
Incidence, etiology, and economic impact
Clinical manifestations
Musculoskeletal impairment
Orthopedic deformities.
Osteoporosis.
Neurological impairment
Hydrocephalus.
Chiari malformation.
Association pathways.
Hydromyelia.
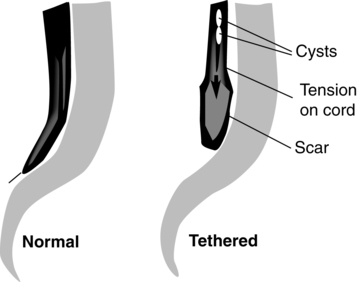
Tethered cord.
Bowel and bladder dysfunction.
Cognitive impairment and learning issues.
Integumentary impairment
Growth and nutrition
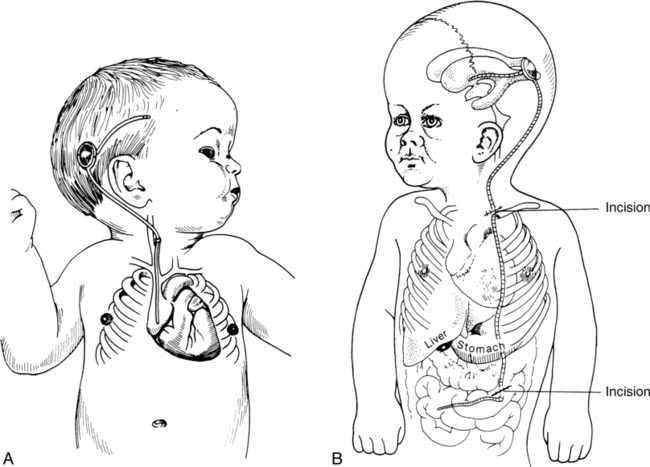
Medical management
Neurosurgical management

Infants
Bulging fontanelSwelling along the shunt tract
Prominent veins on scalp
Downward eye deviation (“sunsetting”)
Vomiting or change in appetite
Irritability or drowsiness
Seizures
High-pitched cry
Toddler
HeadacheVomiting or change in appetite
Lethargy or irritability
Swelling along the shunt tract
Seizures
Onset of or increased strabismus
Older child
All the above, plus:Deterioration in school performance
Neck pain or pain over myelomeningocele site
Personality change
Decrease in sensory or motor functions
Incontinence that begins or worsens
Onset of or increased spasticity
Urological management
Related posts:
 Psychosocial aspects of adaptation and adjustment during various phases of neurological disability
Complementary and alternative therapies: beyond traditional approaches to intervention in neurological diseases and movement disorders
Contemporary issues and theories of motor control, motor learning, and neuroplasticity
Electrophysiological testing and electrical stimulation in neurological rehabilitation
Neuromuscular diseases
Pain management
Psychosocial aspects of adaptation and adjustment during various phases of neurological disability
Complementary and alternative therapies: beyond traditional approaches to intervention in neurological diseases and movement disorders
Contemporary issues and theories of motor control, motor learning, and neuroplasticity
Electrophysiological testing and electrical stimulation in neurological rehabilitation
Neuromuscular diseases
Pain management
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Spina bifida: A congenital spinal cord injury




Only gold members can continue reading. Log In or Register to continue
