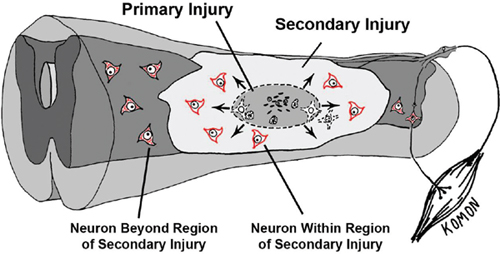
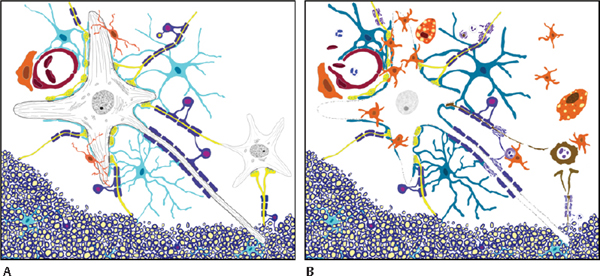
33 Key Points 1. CNS injury induces a cascade of overlapping degradative and reparative processes that interact to determine the final functional outcome. 2. Targeting these processes for therapeutic purposes will be complicated by differential responses of individual cell types and the time course over which they respond. 3. Sensitive and reliable preclinical models are critical for translation of therapies. 4. Several promising pharmacological treatments are now available based on preclinical studies. Edema and excitotoxic cell death have long been targets for therapies aimed at reducing secondary damage after central nervous system (CNS) injury.1–3 Recently, it has become clear that acute inflammatory reactions to injury interact with, and may drive, some aspects of secondary injury.4 The concept of secondary injury is key to the search for acute therapies, although details of the complex cascade of secondary events in spinal cord injury (SCI) and other acute neural damage are lacking. It is generally thought that, in SCI, a frank mechanical disruption of cellular membranes, including breakage of endothelial cells and the release of peripheral blood products into the cord, is followed by the centripetal spread of injury mediated by the biochemical and biological consequences of this initial damage1,3,5 (Fig. 33.1). But the relative roles of extracellular glutamate, blood products released from the injury site, inflammation, and oxidative stress are really not well understood, although each has been the target of intensive investigations. In addition, injury initiates reparative processes as well as degradation, and it is often difficult to differentiate the “positive” from the “negative” biological events after injury. One way to conceptualize this complexity is to view secondary injury and repair in terms of the cell–cell interactions that occur between the multiple cellular constituents of the injured spinal cord or brain (Fig. 33.2). Although each of the cellular constituents of the cord can be identified as a group of phenotypically related cells (e.g., glia, neurons, endothelia), they share many biological response elements, including common neurotransmitter and cytokine receptors, protective mechanisms aimed at reducing cellular stress, and effectors (e.g., glutamate and cytokine release) that make targeting isolated aspects of the injury process problematic. Fig. 33.1 Schematic of the secondary injury process after spinal cord injury. Mechanical impact to the cord concentrates energy in the central region where blood vessels and cell membranes are disrupted immediately. Products of that destruction include glutamate and cytosolic contents that are thought to produce a wave of cell death moving outward from the primary lesion. Cells on the periphery of the expanding lesion are in danger of excitotoxic cell death due to the effects of cytokines and reactive oxygen (see Fig 33.2 and legend), and are the targets for pharmacological treatments aimed at increasing sparing. This chapter discusses several new, promising therapeutic strategies aimed at secondary injury in the context of the interaction between neurons, astrocytes, oligodendrocytes, and their precursor cells (OPCs), microglia and vascular elements, and how those effects may be translated into alterations in the response to injury characterized by edema, excitoxicity, and acute inflammation. The classic example of this is, of course, methylprednisolone (MP), which is a steroid with high activity at glucocorticoid receptors. Its multiple mechanisms of action, however, are thought to include reduction of both edema and lipid peroxidation.6–8 Further, there is evidence that MP reduces endothelial cell damage, thus promoting vascular stability and blood flow,9,10 that MP is antiinflammatory,8 and that MP can reduce axonal retraction after SCI.10 Because there are extensive cell–cell interactions between the multiple cell types affected by SCI, and because many of the potential targets of therapies are shared between multiple elements of secondary injury, it is useful to consider how promising therapies may interact with multiple targets. This is the strategy behind looking for “dirty drugs” that might be most useful in reducing secondary injury.1 It is also worth noting that the drugs that might reduce secondary injury might also affect the endogenous reparative responses of the cord to injury. For example, reduction of the acute inflammatory response to injury by MP has also been associated with a reduction in the production of neurotrophic factors or progenitor cell proliferation that might aid in repair.11–13 Although MP meets many of the criteria of a promising dirty drug, and there is some evidence supporting its promise in rodent models,14 there have been continued failures in attempts to show consistent effects in rodent contusion models of SCI.15 Indeed, it has been difficult to find any treatments aimed at secondary injury that show consistently positive effects in multiple laboratories.16,17 This is likely due, at least in part, to the complicated multiple and redundant pathways leading to tissue damage after injury. Fig. 33.2 A cartoon of the cellular constituents (A) before and (B) after injury. The central importance of the “neurovascular unit” (NVU) in the secondary injury response should be emphasized. Endothelial cell breakdown leads to extravasation of blood and plasma, which contain fibrin, and which can result in vasogenic and cytotoxic edema, leading to swelling of astrocytes, vascular stasis, and continued blood-brain barrier disruption. These events can induce microglial activation, which releases a host of cytokines, including the proinflammatory cytokine tumor necrosis factor-α (TNF-α). TNF can induce the release of glutamate from astrocytes as well. This, in combination with release of glutamate from damaged axons and neurons, can depolarize intact neurons, leading to excess Ca2+ influx via N- methyl-d-aspartate (NMDA) receptors. Excess TNF can move Ca2+ permeable α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid (AMPA) receptor to the neuronal surface, thus increasing the CA2+ load on surviving neurons. All of the resulting cellular death releases intracellular molecules that can in turn activate microglial cells and astrocytes further through, for example, the activation of purinergic receptors and Toll-like receptor 4 signaling. The resultant reactive oxygen species (ROS), along with cytokines, can attack nearby oligodendrocytes, which also respond to glutamate through AMPA receptors, resulting in acute or delayed oligodendrocyte death and demyelination of intact axons. At the same time, glial responses to injury can be protective, including the rapid removal of glutamate from the extracellular space by astrocytic glutamate transporters, and the production of neurotrophic and neuroprotective molecules by both astrocytes and microglia. In addition, injury induces the proliferation and differentiation of endogenous progenitor cells, including those characterized by surface expression of the proteoglycan NG2. In more chronic phases after injury, the result of all these interactions may be the deposition of glial scar and extracellular matrix molecules that retard axonal growth. Even this rather long list of events is really just a snapshot of the complexity inherent in the response of the spinal cord parenchyma to injury. Each of these events represents a target for therapies. (Astrocytes, turquoise; apoptotic neuron and degenerating axons and terminals, brown; normal axons and terminals, yellow; endothelial and red blood cells, red; microglia and macrophages, orange; pericyte, orange; neurons, gray; oligodendrocytes, blue with magenta nuclei and myelin, blue; polymorphonuclear leukocytes,– white with blue nuclei.) Figure 33.1 shows the central hemorrhagic injury due to breakage of blood vessels by trauma. However, vascular damage appears not to be a single mechanical event, but rather is part of the injury cascade can also proceed over time. This has been appreciated in earlier studies of blood flow and vascular status,18,19 but recent attention has been placed on a dynamic expansion of secondary hemorrhage. Progressive hemorrhagic necrosis appears to be related to active breakdown of vascular endothelial cells through a process of endothelial cell swelling and oncosis that is mediated by sulfonylurea receptor 1 (SUR1)-regulated NC (Ca-ATP) channels and transient potential cation channel subfamily M member 4 (TRPM4) channels expressed in response to injury and hypoxia.20,21 SCI induces upregulation of both SUR1-regulated channels and Trpm4. Blockade of SUR1-regulated channel expression, or receptor blockade using glibenclamide or other drugs, produces remarkable reduction in progressive hemorrhagic necrosis. Trpm4 -/- mice have much attenuated vascular breakdown after a cervical contusion injury.20 Reductions of channel expression or activity have marked effects on injury progression and appear to improve outcome substantially.20,21 The prevention of progressive vascular disruption and protection of endothelial cells presumably blunts all of the downstream complex sequelae, including glutamate and adenosine triphosphate (ATP) production, microglial activation, and cytokine production, and the progression of cell death that ensues, including the longer-term effects on oligodendrocyte apoptosis.22,23 It is less clear how this endothelial cell progression of damage may affect injuries that are less mechanical and more ischemic in origin, but SUR1-regulated channels are also implicated in the progression of hemorrhagic necrosis seen in ischemic stroke models.24 Disruption of the blood–brain barrier is therefore linked to endothelial cell edema, and the critical importance of the “neurovascular unit” in secondary injury is emphasized.25 In this regard, it is interesting that hypertonic saline, which is thought to provide protection for endothelial cells by reducing cellular swelling,26 has been shown in our laboratory to reduce magnetic resonance imaging indices of edema and hemorrhage27 in the first 8 hours after a cervical contusion injury model similar to that used to demonstrate the positive effects of endothelial cell protection by blocking SUR1-regulated NC-Ca-ATP channels or Trpm4 already described. For example, dysregulation of the Trpm4 channels results in unchecked Na+ influx, swelling, and oncosis of endothelial cells, as well as astrocytes and neurons.20 In turn, release of ATP by dying cells can play a role in the production of secondary injury. This has been highlighted by recent work in which blockade of ATP activation of P2X7 receptors has been shown to improve outcomes after experimental SCI in rats.28,29 Wang et al.28 provided an elegant demonstration of the release of extracellular ATP after SCI using luciferase-mediated breakdown of luciferin in the presence of ATP to image ATP release in vivo after a mechanical injury to the dorsal spinal cord. After laminectomy, a charge coupled device (CCD) camera and in vivo microscopy were used to show the spread of luciferase-mediated fluorescence after a mechanical injury to the surface of the exposed cord. Concentrations of ATP were estimated qualitatively, but the experiment clearly showed a progression of ATP release over time, yielding another confirmation of the concept of secondary injury spread. When ATP activation of P2X7 receptors was blocked by OxATP, cell death was reduced and recovery was enhanced. Treatments reduced apoptosis after a thoracic contusion injury in rats, as measured in a TUNEL assay, and also dramatically improved Basso, Beattie, Bresnahan (BBB) locomotor scores30 at 6 weeks postinjury from ∼ 9 to 14, corresponding to a difference between weight support and no or little stepping, to consistent plantar stepping and consistently coordinated locomotion in the treatment group. In a follow-up study,29 P2X7 receptor blockade was obtained using a novel antagonist, brilliant blue G (BBG), a Coomasie dye analogue of the food coloring additive FD&C blue dye no. 1. Even large doses of this dye are not toxic, and indeed, the authors point out that humans consume massive quantities of this food coloring every day. Thus it was particularly intriguing that this treatment also had significant effects on outcomes from thoracic contusion injuries in rats. BBB locomotor scores were improved from an average of 9.4 to 11.9 at 6 weeks, corresponding to a difference between weight support and no or little plantar stepping in untreated subjects and consistent plantar stepping with some coordinated locomotion in the treatment groups. BBG treatments of 10 or 50 mg/kg were also found to improve tissue sparing at the lesion site, and, importantly, to reduce microglial activation at early times after injury (as measured by immunocytochemistry and microglial morphology). In addition, BBG treatment reduced the infiltration of macrophages, neutrophils, and CD8-positive T-cells. All of these results are consistent with a powerful antiinflammatory effect mediated by blockade of microglial activation through the P2X7 receptor. But, in addition, neurons (and to a lesser extent astrocytes) also have P2X7 receptors. Because ATP can induce activity and excitotoxicity in neurons as well, the BBG treatment also represents a multiple-cell targeted therapy. If this work can be replicated, it should serve as an important stimulus for more secondary injury treatments aimed at early stages in the inflammatory cascade. This work supports the use of antiinflammatory strategies, even though the reports of efficacy of antiinflammatory drugs like minocycline have shown mixed positive and negative effects.17,31,32 Glutamate reaches toxic levels minutes after SCI,33 and drugs that reduce the activity of glutamate receptors, allowing Ca2+ influx, have been used for many studies of secondary neuroprotection. However, the most effective agents, including MK-801, tetrodotoxin (TTX), and others,34–36 are not suitable for clinical use. Recently, increased attention has been given to neuroprotective drugs that are used as antiepileptic agents, including phenytoin and riluzole. These drugs also share multiple mechanisms of action, including reduction of Na+ influx to ligand and voltage-gated channels,37 and reduction of glutamate release. Studies in models of epilepsy also show reduced cellular damage and excitotoxicity.38 In addition, riluzole has been tested in human clinical trials for amyotrophic lateral sclerosis (ALS), with some efficacy demonstrated.39,40 In rat SCI models, antagonists of glutamate receptors, including the AMPA/kainate receptor antagonists NBQX34 and GYKI 52466,41 have shown some positive effects on behavioral recovery (BBB score outcomes of ∼ 2 points difference). Riluzole, typically used as an anticonvulsant, has shown rather impressive neuroprotection and amelioration of neurological deficits in a rat clip-compression model of SCI.37 Interestingly, several other compounds that are thought to share the Na+ channel blocking properties of riluzole (e.g., phenytoin) failed to show efficacy in the same study. Another study using a more rapid contusion injury model, also in rats, showed efficacy only when riluzole was given in combination with MP.42 Riluzole’s actions might also be due to its apparent reduction of glutamate release by astrocytes.43 Riluzole is currently in clinical trials for SCI and has also shown efficacy in trials for ALS.39,40 Riluzole also has activity in reducing glutamate release43 and thus affects multiple mechanisms of action against secondary injury. Again, multiple targets are better, but it is difficult to predict efficacy in rodent models based on mechanisms of action alone. Importantly, the actions of glutamate on oligodendrocytes and oligodendrocyte progenitor cells via AMPA-receptor-mediated cell death and apoptosis are also critical in secondary injury.3 Indeed, many of the drugs that show some efficacy against neuronal excitotoxicity also have been shown to inhibit secondary oligodendrocyte cell death and demyelination.32 Finally, we turn to the role of proinflammatory cytokines in secondary injury. The tetracycline derivative minocycline has antiinflammatory properties and has been used to reduce microglial activation in an attempt to reduce the production and release of proinflammatory tumor necrosis factor-α (TNF-α), for example, which rise precipitously after SCI.44 Proinflammatory cytokines in turn can activate microglia as well as increase glutamate release from astrocytes.45 Although a direct “bystander effect” has been postulated for both neurons and oligodendrocytes,46 recent work in our laboratory and others has highlighted an interactive effect between cytokine actions and glutamate-induced excitotoxicity. Thus, TNF-α greatly exacerbates the excitotoxic effects of kainic acid nano-injected into the spinal cord gray matter.47 This effect is completely blocked by the AMPA/kainic acid receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-drone (CNQX). Thus TNF has an amplifying effect on glutamate signaling through AMPARs. This amplifying effect was shown in vitro to be due to a specific TNF-α effect on the trafficking of AMPARs to the surface of the neuronal membrane.48 This increased the effects of AMPAR-mediated cell death, and the death was blocked by drugs that reduced PI3K activity and thereby the TNF-induced increases in trafficking.49 Contusion SCI also could be shown to increase the surface localization of AMPARs that lacked GluR2.50 Such AMPARs allow passage of Ca2+ ions, which are normally blocked by the presence of the GluR2 subunit. Thus more GluR1-containing and fewer Glur2-containing receptors on the surface would lead to increased excitotoxic effects of the same amount of extracellular glutamate. This was confirmed by using a soluble TNF receptor protein 1 (sTNFr1) to sequester injury-induced TNF. This treatment reduced surface expression of GluR2-lacking AMPARs and also reduced neuronal cell death after SCI.50 Despite the frustrations of numerous failures to show consistent positive outcomes using drugs aimed at secondary injury in CNS trauma and stroke,1 the exciting results of the selected positive outcomes reviewed in this chapter suggest that the prospects for better therapies for secondary injury after SCI seem promising indeed. Growing understanding of the roles of multiple destructive and reparative pathways under the control of complex cell–cell interactions is providing more insight into how drugs targeted to “nodal” mechanisms might be effective. Further, it is clear that combinatorial therapies are likely to be needed, as has been the case in pharmacological approaches to multiple treatment targets in cancer therapeutics.1 It does seem clear that the concept of an evolving secondary injury after SCI is holding up to continued scrutiny, and that these events evolve over a long enough time to provide a window of therapeutic opportunity. The results of human trials under way, for example riluzole,51 will hopefully pave the way for continued efforts aimed at this elusive target. Pearls
Promising Preclinical Pharmacological Approaches to Spinal Cord Injury
 Therapies Aimed at One Target May Affect Multiple Aspects of Secondary Injury
Therapies Aimed at One Target May Affect Multiple Aspects of Secondary Injury
 Blood Vessel Injury Is a Point of First Defense
Blood Vessel Injury Is a Point of First Defense
 Cellular Energy Loss and Depletion of ATP Appear to Be Important in Cellular Dysregulation after Injury
Cellular Energy Loss and Depletion of ATP Appear to Be Important in Cellular Dysregulation after Injury
 ATP Activation of Microglia and Neurons, As Well As Cell Death–Induced Release of Intracellular Stores of Glutamate, Contribute to Excitotoxicity 33
ATP Activation of Microglia and Neurons, As Well As Cell Death–Induced Release of Intracellular Stores of Glutamate, Contribute to Excitotoxicity 33
 Proinflammatory Cytokines Play an Important Role in the Secondary Injury Cascade
Proinflammatory Cytokines Play an Important Role in the Secondary Injury Cascade
 Conclusion
Conclusion
 The complex interactions between the multiple cellular constituents of the nervous system (glia, neurons, endothelial cells) present in the neuropil determine the outcome of injury, and the cascade of events that ensues includes both degradative and reparative processes.
The complex interactions between the multiple cellular constituents of the nervous system (glia, neurons, endothelial cells) present in the neuropil determine the outcome of injury, and the cascade of events that ensues includes both degradative and reparative processes.
 The proinflammatory cytokine, TNF, rapidly released in response to injury, exacerbates the excitotoxic effect of injury-induced release of glutamate by increasing the surface expression of AMPA-type glutamate receptors. This same process is involved in the physiological regulation of synaptic strength. Thus injury hijacks a normal physiological process to produce an injury cascade.
The proinflammatory cytokine, TNF, rapidly released in response to injury, exacerbates the excitotoxic effect of injury-induced release of glutamate by increasing the surface expression of AMPA-type glutamate receptors. This same process is involved in the physiological regulation of synaptic strength. Thus injury hijacks a normal physiological process to produce an injury cascade.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


