Osteonecrosis and Bone Marrow Edema Syndrome
David R. Marker, Thorsten M. Seyler, Michael A. Mont and Edward F. McCarthy
Key Points
Introduction
Osteonecrosis of the hip (ON) and bone marrow edema syndrome (BMES), also called transient osteoporosis of the hip (TOH), are causes of hip pain in middle-aged patients who show evidence of bone morrow edema. Although some literature has suggested that BMES may be a reversible form of osteonecrosis rather than a disease entity of its own,1,2 no definitive documentation is available to support this theory. The current general consensus is that BMES is a unique disease entity.3 In contrast to ON, BMES is a transient but painful condition of the hip that is limited to marrow changes without associated joint space narrowing and arthritic changes. Because of the low incidence of BMES, few reports have described large series, making it difficult to further elucidate the pathophysiology of this disease. It was originally defined by Curtiss and Kincaid in 1959 as a syndrome of transient demineralization of the hip in the third trimester of pregnancy.4 It has since been found to occur more frequently in young and middle-aged men.5–7 In contrast to the extremely low incidence of BMES, osteonecrosis is a more common condition that has existed for millennia; evidence of the disease has been found in Egyptian mummies.8 ON is defined by compromise of bone blood circulation that leads to ischemic death of subchondral bone in the femoral head. The hallmark that separates these two entities is that most patients who are diagnosed with ON progress to advanced stages of the disease with femoral head collapse and painful arthritis that require total hip arthroplasty, in contrast to the usual self-limited nature of BMES (Fig. 35-1).
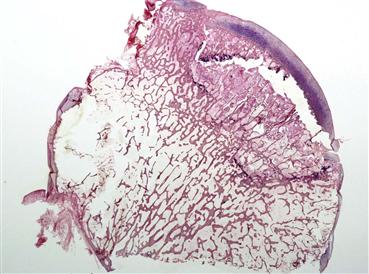
Figure 35-1 This macro-photomicrograph of a hip specimen demonstrates the subchondral fracture that corresponds with the “crescent sign” seen on radiographs in advanced-stage osteonecrosis. Patients who reach this stage of disease invariably require reconstructive total joint surgery. Because this degree of joint destruction does not occur in patients who have bone marrow edema syndrome, it is important that surgeons are able to distinguish these disease entities and provide appropriate treatment.
Differentiation of ON and BMES is complicated by various terms that have been used to describe these two diseases. Ultimately, it is important for orthopedic surgeons to understand this terminology so they can properly use the current literature in their diagnoses, decision making, and surgical management. Some of the descriptions that have been used are based on the underlying pathogenesis or the clinical nature of the disease, but others are outdated and are no longer commonly used. Lequesne and associates were the first to characterize the syndrome of transient demineralization of the hip as transient osteoporosis.5 The usefulness of transient osteoporosis of the hip as a clinical term supplanted use of the original terminology of transient demineralization of the hip. In general, many investigators consider BMES and transient osteoporosis of the hip as the same disease. Because of its similarity to other transient clinical entities at other sites in the body, it is now included in the umbrella of diseases commonly referred to as bone marrow edema syndromes.9 In cases of bone marrow edema characterized by concurrent or subsequent involvement of additional anatomic sites other than the primary hip, the overall syndrome may be referred to as regional migratory osteoporosis. Although it remains unclear whether regional migratory osteoporosis is the same entity as is seen in cases of isolated BMES that have no other sites of involvement, many clinician experts consider regional migratory osteoporosis a variant of BMES.10
Other authors initially postulated that BMES was a form of reflex sympathetic dystrophy (now called complex regional pain syndrome, type 2), but this theory has largely fallen out of favor.11,12 Also, as previously noted, some authors have proposed that BMES may be a nonprogressive, reversible form of osteonecrosis.1 Regardless of the proposed pathophysiology, BMES remains the most commonly used term to describe this disease. In the case of ON, the terminology has been less variable. Initially, avascular necrosis was commonly used. This term was an accurate descriptor of this disease for cases with a known traumatic origin in which the vascular supply to the femoral head was directly compromised. However, this terminology was deemed inaccurate in its use when describing lesions in individuals who had associated risk factors such as corticosteroid usage, systemic lupus erythematosus, or Gaucher’s disease, where bone marrow displacement and increased compartment pressure were considered to be the likely pathogenetic mechanisms. Osteonecrosis has subsequently become the preferred term. Although ON can be grouped with other diseases that cause bone marrow edema, it is typically studied as a distinct entity.
Part of the difficulty in differentiating ON from BMES may be that patients with these diseases can present with seemingly nonspecific groin pain and equivocal findings on physical and radiologic examination. Nevertheless, some progress has been made in understanding the underlying origins of these conditions and in examining the outcomes of various surgical and nonsurgical treatments. The present work will serve as a review of current knowledge about these conditions to assist clinicians in their management. This chapter begins by discussing the differences in epidemiology, risk factors, pathophysiology, and clinical and diagnostic features that can be used to distinguish these two diseases, while providing some of the arguments that have been put forth against this taxonomy. The second part of the chapter reviews various treatment options available for each condition and provides a discussion of their rationale and indications, along with a summary of results obtained when various techniques were used.
Epidemiology and Risk Factors
The exact prevalence of ON and BMES remains undefined. It is estimated that approximately 10,000 to 20,000 new cases of ON are diagnosed in the United States each year.13 In more than 10% of these cases, the disease will also involve the knee and the shoulder, and in less than 3% of patients, the disease will be multifocal and will affect more than three anatomic sites. These numbers are clinically more important when it is considered that more than 10% of hip arthroplasties performed in the United States are related to ON. The prevalence of BMES is considerably less than that of ON. Only several hundred cases of BMES have been reported in the literature, and the incidence is less than 1% of the number of ON cases each year.14
The prevalence of ON is higher in specific at-risk populations. Recent studies have evaluated the increased prevalence of ON associated with corticosteroid use, alcohol abuse, sickle cell disease, and genetic factors. Griffith and associates reported that 5% of patients (12 of 254) with systemic adult respiratory syndrome (SARS) first had evidence of osteonecrosis of the femoral head, and that the cumulative prednisolone-equivalent dose was the most important risk factor, with the risk being 0.6% for patients receiving a dose less than 3 g, and 13% for those receiving a dose greater than 3 g. For transplant patients on high doses of corticosteroids, the prevalence of ON has been reported to be between 3% and 23%.15–17 Recognition of the association between ON and corticosteroids has led to preventative measures, and organ transplant patients on modern immunosuppressive drugs have a risk of ON that is likely at the lower end of the spectrum previously reported—near 5%.18 The association of alcohol abuse with increased risk for ON was evaluated in separate studies by Hirota and associates and Matsuo and colleagues. Investigators found similar findings with a clear dose-response relationship. Hirota and coworkers reported a higher risk for development of ON in occasional drinkers (<8 mL of alcohol once a week, but not daily; relative odds = 3.2) and in regular drinkers (≥8 mL of alcohol daily; relative odds = 13.1) than in controls. The dose-response relationship reported in their study revealed relative odds for current drinkers of 2.8, 9.4, and 14.8 in association with ethanol intakes of <320, 320 to 799, and ≥800 g/wk, respectively. Matsuo and associates reported an elevated risk for regular drinkers (>8 mL of alcohol every day; relative risk = 7.8). They also described a dose-response relationship, with relative risks of 3.3, 9.8, and 17.9 for current drinkers consuming <400, 400 to 1000, and ≥1000 mL/wk of alcohol, respectively. The incidence of ON in sickle cell patients was recently studied in a cohort of 200 patients over a mean 15-year follow-up.19 Osteonecrosis was greatest among groups with the SS genotype (43% developed multifocal disease), followed by those with the hemoglobin SC genotype (38%) and the Sβ+ thalassemia genotype (19%).
Genetic factors that may predispose individuals to ON remain poorly defined. Familial high plasminogen activator inhibitor and resulting hypofibrinolysis were initially associated with the development of ON by Glueck and colleagues,20 who reported that, compared with control subjects, patients with osteonecrosis were more likely to have heterozygosity and homozygosity for the hypofibrinolytic 4G polymorphism of the plasminogen activator inhibitor-1 gene. Other studies have reported that genes affecting lipid transport and metabolism,21 or production of increased catalase and decreased nitric oxide,22,23 may increase the risk for ON.
Much less is known regarding the risk factors for BMES. Pregnancy was the first risk factor identified. Even in patients who are believed to be at risk, the incidence of BMES is low. It has been reported that middle-aged men are at higher risk.24 However, early diagnosis of pregnant BMES patients remains important, because evidence suggests that they have a unique risk for femoral neck and stress fractures compared with their nonpregnant counterparts. Pregnant patients who develop BMES should be followed clinically until radiographic evidence (MRI) indicates that the hips have undergone adequate reconstitution of their bone mass.25
Pathogenesis
Although ON is a well-described clinical entity, its cause and pathogenesis have not been completely elucidated. The pathogenesis of BMES is even less well defined. In most cases, the cause of ON can be characterized by a final common pathway of (1) focal intravascular coagulation and subsequent thrombosis that affect the terminal arterioles or the postsinusoidal venules, and/or (2) increased intraosseous pressure that is postulated to compress the subchondral microvasculature. The predilection of the femoral head for coagulation and thrombosis may be based on the microanatomy of its blood supply. Minimal collateral circulation is seen, and end arterioles form vascular arcades that must make abrupt turns at the ends of cortical bone before returning venous blood from the femoral head.
The pathogenetic mechanism of osteonecrosis can also be characterized according to underlying causes or associated risk factors. ON is a multifactorial disease that is associated with various direct and indirect risk factors. Direct causes include trauma, Caisson’s disease, chemotherapy, Gaucher’s disease, and radiation. Indirect causes that have been associated with ON include alcohol and smoking abuse, coagulation abnormalities (thrombophilia, hypofibrinolysis), corticosteroid use, inflammatory bowel disease, organ transplants, pregnancy, and systemic lupus erythematosus.
In cases such as trauma or radiation, the pathogenetic mechanism directly causes necrosis. Other causes initiate events that lead to thrombosis and disruption of the microcirculation of the femoral head. For example, in sickle cell disease and Caisson’s disease, direct restriction or occlusion of blood vessels is noted. Other factors that may contribute to risk in sickle cell patients include higher blood viscosity and bone marrow hyperplasia. Additionally, deformed erythrocytes may cause microinfarcts in the subchondral bone.26 In patients who develop osteonecrosis after corticosteroid use or alcohol abuse, intraosseous pressure is increased likely as the result of enlarged adipocyte size and proliferation. In addition, fat emboli may become trapped in the end arterioles and occlude these vessels in the subchondral bone. Subsequently, damage to the endothelium initiates the clotting cascade, and the microvasculature is compromised. Gaucher’s disease, leukemia, and myeloproliferative disorders are thought to increase pressure in the bony compartments of the hip by displacing intraosseous marrow in the bony compartments of the femoral head and neck. Because the bone marrow cannot expand, the bone involved cannot compensate for increased pressure; this leads to collapse of vessels, ischemia, and cell damage. Systemic lupus erythematosus has been shown to be an independent risk factor in osteonecrosis. Recent reports have assessed whether patients who have systemic lupus erythematosus may be at increased risk for osteonecrosis if they also present with Raynaud’s phenomenon, hyperlipidemia, and/or high levels of anticardiolipin or antiphospholipid antibodies, but additional studies with more patients are needed to further assess any correlation.27,28 Controversy concerning the influence of antiphospholipid antibodies is ongoing, with some studies suggesting that there is an association,29 while others suggest there is not.30 Much recent research has been undertaken to explore the risk of development of osteonecrosis in patients who have inherited coagulation disorders. Associations have been shown between ON and thrombophilia and hypofibrinolysis because of an increase in blood clots and a decreased ability to lyse blood clots, respectively.31–35
The cause of BMES is unknown. Mechanisms such as demineralization, neurogenic compression, reflex sympathetic dystrophy, and obstruction of venous return with localized hyperemia have been proposed previously but are no longer widely accepted.4,5,11,12 Currently, two pathogenetic mechanisms propose that BMES (1) results from sequelae of a subchondral insufficiency fracture at, or near, a weight-bearing surface,36 or (2) is caused by subacute transient ischemia seen along the spectrum of osteonecrosis.9 The postulate that a subchondral insufficiency fracture is associated with BMES is supported by recent research assessing regional accelerated phenomenon activation. When bone is exposed to noxious stimuli such as microfractures, it has been shown to undergo localized modeling and remodeling at rates of up to 10 times normal. Prolonged activation of this phenomenon may result in transient osteoporosis.37 Imaging studies suggest that subchondral insufficiency fractures may eventually be detected in all BMES patients as the resolution and technology of advanced MRI continue to improve.36,38
Histopathology
Some of the histologic features of ON and BMES are similar. It has been suggested that changes such as marrow edema, dilated medullary sinuses, and fat vacuoles in the interstitial fluid are indicative of a common pathophysiologic entity.39 Hofman and associates suggested that BMES is a precursor to osteonecrosis than can progress to advanced osteonecrosis or can improve if ischemic levels are subcritical.40 However, key differences have been noted in the histopathologies of ON and BMES, the most notable being the continued presence of osteocytes in BMES. This finding is used in the argument that these are distinct entities.
The histopathologic process of ON is dynamic and nondiscrete but can be defined in terms of four key stages (Box 35-1). The first stage is marked by true necrosis with morphologic changes that occur in tissues after death (Fig. 35-2). These histologic changes do not begin until several days after death. Therefore, histologic study of bone immediately after an infarct will reveal no changes. The first change to occur is dissolution of hematopoietic cells. This begins approximately 2 to 3 days after death. Then, osteocytes drop out of lacunae anywhere from 2 days to 4 weeks after death. Marrow fat begins to show necrotic changes about 5 days after the bone is dead. Therefore, bone may not be recognizably dead by histologic study until 5 or more days after irreversible cessation of cellular activity. These early histologic changes correlate with Ficat stage I.
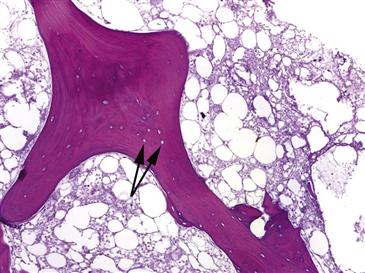
Figure 35-2 This histopathologic sample represents the first stage of osteonecrosis: cell death and necrosis. Bone death is evident by the absence of osteocytes in the trabecular bone lacunae (black arrows). The surrounding marrow is necrotic with almost complete absence of hematopoietic elements.
After several weeks following tissue death, necrotic marrow begins to show dystrophic calcification (Fig. 35-3). This is a common morphologic change in fat necrosis in any location of the body. After several weeks, a reparative reaction begins at the margin of the bone infarct. Ingrowth of granulation tissue brings multipotential cells that begin to lay down new viable bone on dead trabeculae. This causes marked thickening of bone trabeculae in a process called appositional new bone formation. As time progresses, this front of reparative bone extends into the center of the infarct; this correlates with radiographic changes of radiodensity (Ficat stage II).
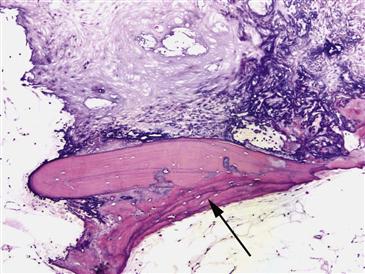
Figure 35-3 This photomicrograph demonstrates the reparative reaction that occurs in Ficat stage II osteonecrosis of the hip (ON). Granulation tissue at the margin of the bone infarct brings multipotential cells that begin to lay down new viable bone (black arrow) on the dead trabeculae. The surrounding fat in this sample has undergone focal dystrophic calcification changes.
The next change that occurs is due to osteoclastic resorption at the viable/necrotic interface. Osteoclastic resorption begins to weaken trabeculae, and a subchondral fracture often occurs. This signals failure of the surface of the bone. It is visible histologically as a crack beneath the articular cartilage and radiographically as a “crescent sign” (Ficat stage III).
The final stage (Ficat stage IV) is the development of osteoarthritis. Histologic features are consistent with osteoarthritis in showing diminution of superficial chondrocytes and proliferation of deeper chondrocytes in broad capsule clusters, with capillary buds penetrating the layer of calcified cartilage in areas overlying the segment of bone that underwent necrosis and subsequent subchondral fracture.
The earliest change in BMES is a change in the marrow (Fig. 35-4). The marrow is filled with a faintly eosinophilic material that is edema fluid. This corresponds radiographically to a bright signal on T2-weighted MRI images (Box 35-2). Mild fibrosis may be associated with vascular congestion. After about 3 weeks, teams of osteoclasts are activated and begin to resorb bone (Fig. 35-5). This will account for the changes of osteoporosis seen in cases that are several weeks old. Resolution of osteoporosis is signaled by ingrowth of reparative tissue with an osteoblastic lining of trabeculae (Fig. 35-6). Osteoblasts are able to deposit new reparative bone on the thinned trabeculae. This process accounts for the resolution of transient osteoporosis.
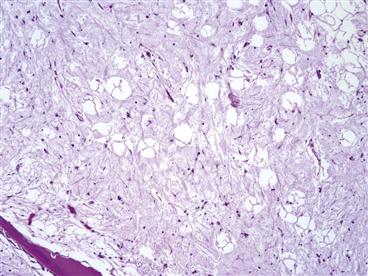
Figure 35-4 The histopathologic finding characteristic of bone marrow edema syndrome is edema in the fatty marrow. This slide demonstrates this edema as evidenced by amorphous material between fat cells.
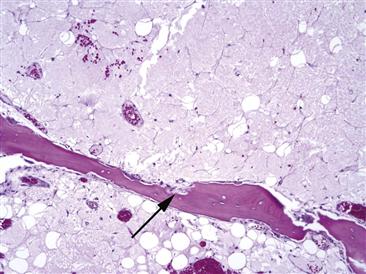
Figure 35-5 As bone marrow edema syndrome (BMES) progresses, osteoclasts begin to form resorption cavities (black arrows). The degree of osteoporosis noted in the hip of the patient is a function of how extensive this resorptive process removes trabecular bone.
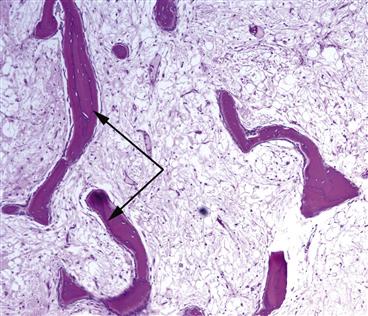
Figure 35-6 Seams of new woven bone (black arrows) are evident in this image of a sample taken from a patient with bone marrow edema syndrome (BMES). This osteoblastic activity restores trabecular bone in the hip back to pre-BMES levels.
Clinical Features and Diagnosis
Patients who have BMES may present with clinical symptoms that are similar to those experienced by ON patients. In both diseases, patients may have disabling hip pain with no known antecedent trauma (with the exception of trauma-associated osteonecrosis of the hip). The pain can be located in the inguinal area, buttocks, or anterior thigh. It is deep and throbbing, worse with ambulation, and marked at night. The patient may have a limp with pain on weight bearing and a positive Trendelenburg sign. Physical examination may elicit nonspecific hip pain on extremes in range of motion that may limit the ability of the patient to move through a full range of motion. The most severe pain is felt in abduction and internal rotation. However, many patients with ON or BMES present with no pain. Patients with these diseases experience a variety of different symptoms, and their diagnosis should be made with the use of MRI.
Several clinical differences can be used to help differentiate between patients with ON and those with BMES (Table 35-1). Although the diseases are similarly present in middle-aged patients and may affect pregnant women,41,42 BMES rarely presents in women other than those who are in their third trimester of pregnancy. Evidence of bilateral disease can be used to exclude a diagnosis of BMES in most cases. However, there are exceptions to these rules, as some evidence suggests that bilateral, as well as early-trimester, BMES may occur on rare occasions.42 In general, although clinical findings may be more suggestive of one of these diseases, physical examination findings are not very helpful in differentiating the two diagnoses; imaging is required.
Table 35-1
Comparison of Osteonecrosis and Bone Marrow Edema Syndrome Clinical Assessments
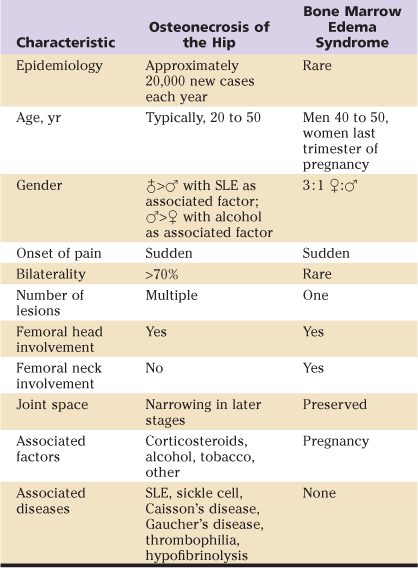
SLE, Systemic lupus erythematosus.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


