Fig. 34.1
Four forms of mutations commonly responsible in oncogenesis: point mutation (a), deletion (b), chromosomal translocation (c), and gene amplification (d)
Name of gene/s | Type of genetic alteration and associated conditions | Decreased/absent protein and function | Tumor with increased risk and related considerations | |
|---|---|---|---|---|
RB | Germline inactivating point mutation or deletion, congenital retinoblastoma | Rb: negative regulator of cell cycle | Osteosarcoma (up to 1,000-fold risk of generation) | |
Soft tissue sarcomas | ||||
P53 | Germline inactivating mutation, Li Fraumeni syndrome | p53: genomic integrity receptor, promotion of DNA repair or apoptosis if needed | Osteosarcoma | |
Pleomorphic rhabdomyosarcoma | ||||
Pleomorphic undifferentiated sarcoma | ||||
INK4a (CDKN2A) | Inactivating small deletions | p16 (a CDK-inhibitor): activation of Rb and regulation of G1/S checkpoint | Osteosarcoma and Ewing sarcoma | |
P14: augmentation of p53 functions | ||||
RECQL4 | Mutation, autosomal recessive inherited Rothmund-Thomson syndrome | ATP-dependent DNA helicase Q4 enzyme: occurrence of abnormalities during DNA replication | Osteosarcoma | |
PTHR1 | Heterozygous function reducing mutations | Reducing the affinity of the receptor for PTH or the receptor expression at the cell surface and subsequent unregulated endochondral ossification | Enchondroma syndromes (Ollier and Maffuci syndromes) | |
EXT1 and EXT2 | Germline loss-of-function mutation | Glycosaminoglycans: regulation of chondrocyte differentiation by providing normal diffusion of Ihh | Sporadic osteochondroma | Disrupted chondrocyte differentiation and osteochondral mass growth |
Hereditary exostosis | ||||
Chondrosarcoma (EXT mutations present in chondrosarcomas secondary to hereditary exostosis) | ||||
APC | Sporadic or germline mutations | Abnormal APC protein and subsequent uncontrolled beta-catenin activity and increased cell adhesion and proliferation | Deep fibromatosis (desmoid tumor) | |
NF1 | Heterogeneous mutations, type I neurofibromatosis | Neurofibromin: negative regulator of the Ras oncogene signal transduction pathway | Neurofibroma | |
Malign peripheral nerve sheath tumor | ||||
IDH1 and IDH2 | Heterozygous somatic mosaic mutations | Modified isoforms of isocitrate dehydrogenase (IDH) enzymes leads to epigenetic changes (hypermethylation of DNA and histones leading to activation of oncogenes and inactivation of anti-oncogenes) | Solitary chondroma and enchondroma | Genetically both normal and abnormal cells are present in these masses |
Enchondroma syndromes (Ollier and Maffuci syndromes) | ||||
Both sporadic and chondromatosis-related chondrosarcomas | ||||
Gene/s | Type of genetic alteration | Mechanisms of effect | Tumor type and related descriptions | |
|---|---|---|---|---|
CTNNB1 | Sporadic mutation | Over-expressed beta-catenin and subsequent increased cell adhesion and proliferation | Deep fibromatosis (desmoid tumor) | |
MDM2 | Amplification | Over-expressed MDM2: inhibition of p53 | Osteosarcoma | |
CDK4 | Amplification | Over-expressed CDK4: inhibition of Rb | Osteosarcoma | |
NF1 | Heterogeneous mutations at q arm of chromosome 17, type I neurofibromatosis | Neurofibromin: negative regulator of the Ras oncogene signal transduction pathway | Neurofibroma | |
Malign peripheral nerve sheath tumor | ||||
MDR-1 | Amplification or translocation | Over-expressed P-glycoprotein (P-gp or ABCB1) and subsequent increased effluxes of structurally diverse compounds from the tumor cell | Various sarcomas (including osteosarcoma), gaining resistance again apoptosis and therapeutic agents | |
Fused EWS and FLI1 genes | t(11;22)(q24;q12) (main alteration present in 85 % of Ewing sarcomas) | Abnormal EWS fusion proteins with multiple effects leading to neoplasia formation | Ewing sarcoma (cells of origin is not clear yet) | |
Fused EWS and ERG genes | t(21;22)(q22;q12) | |||
Fused EWS and ER81 genes | t(7;22)(p22;q12) | |||
Fused EWS and CHN genes | t(9;22)(q22;q12) | Abnormal EWS fusion protein with multiple effects leading to neoplasia formation | Extraskeletal myxoid chondrosarcoma | |
Fused EWS and WT1 genes | t(11;22)(q13;q12) | Desmoplastic small round cell tumor | ||
Fused EWS and ATF1 genes | t(12;22)(q13;q12) | Clear cell sarcoma | ||
Fused COLA1 and PDGF-beta genes | t(17;22)(q22;q15) | Over-expression of PDGF-beta | Dermatofibrosarcoma protuberans | |
Fused USP6 and promoter genes | Rearrangement of chromosome 17p13 | USP6 over-expression and subsequent increased activity of NFKB and matrix metalloproteinases | Primary aneurysmal bone cyst | |
Fused MYH9 and USP6 genes | t(17;22) | Unknown | Nodular fasciitis (proliferating cells have not all hallmarks of cancer and the condition underwent self-limitation) | |
GNAS1 | Somatic gain-of-function mutation | A Gs-protein with constant activity leading to interruption of differentiation of osteoblasts from precursor cells | Fibrous cortical defect | Developmental clinical conditions due to mutations after the formation of the skeleton |
Monostotic fibrous dysplasia | ||||
Fused M-CSF and promoter genes | t(1;2)(p13;q37) | Over-expressed monocyte colony-stimulating factor and subsequent increased macrophage proliferation | Local type tenosynovial giant cell tumor (giant cell tumor of tendon sheath) | |
Diffuse type tenosynovial giant cell tumor (pigmented villonodular synovitis) | ||||
Fused PAX3 and FOXO1 genes | t(2;13)(q35;q14) | Synthesis of associated chimeric transcription factors and subsequent disruption of skeletal muscle differentiation and tumor formation | Alveolar rhabdomyosarcoma | |
Fused PAX7 and FOXO1 genes | t(1;13)(q36;q14) | |||
Fused SS18 and SSX1 genes | t(x;18)(p11;q11) | Synthesis of associated chimeric transcription factors and interruption in the control mechanisms of cell cycle and tumor formation | Synovial sarcoma | |
Fused SS18 and SSX2 genes | ||||
Fused SS18 and SSX4 genes | ||||
Fused FUS and DDIT3 genes | Amplification of 12q13-q15 | Over-expressed MDM2 and subsequent p53 inhibition and arrest of differentiation of adipose tissue cells | Well-differentiated liposarcoma | |
t(12;16)(q13;p11) | Myxoid liposarcoma | |||
Noncoding RNAs are one of two basic mechanisms of gene function regulation in living cell. The other is driven by epigenetic factors and will be described in the next paragraph. Noncoding RNAs are transcribed from DNA segments but not translated to proteins. The main types of noncoding RNAs are micro-RNAs (miRNAs) and long noncoding RNAs (lncRNAs). The main functions of miRNAs are regulation of translation of their target mRNAs into the proteins. Every miRNA modulates multiple protein-coding processes. Posttranscriptional silencing of a gene comes true by base pairing between associated mRNA’s non-translating region and miRNA-multiprotein complex. This pairing process leads to either cleavage or repression of mRNA, ceasing its translation into the protein. It seems that miRNAs have important roles in either non-pathologic or pathologic tissue developments. There are efforts to synthetically produce miRNA-like agents that can bind specific mRNAs. These kinds of agents can be used for diagnostic aims as well as for silencing the oncogenes and treatment of neoplasms. Long noncoding RNAs enhance and silence gene transcriptions by binding to specific regions of DNA chain. To supply these effects, lncRNAs use different mechanisms. Their roles in human diseases including cancers are also under investigation [2, 5, 6].
Epigenetic factors are regulating factors of gene expression that are not originated from DNA. Changes of these factors are also heritable and mostly concern histones. Histones are low molecular weight proteins whose combinations make up a central core for DNA segment envelops. Histones and DNA segment combinations form nucleosomes, the basic structural units of chromosomes. In nucleosomes, the DNA segment is wrapped around histones in changing forms and compactness. These changes occur due to the dynamic and regulating structure of nucleosomes. Proteins and chemical agents can effect on both histones and DNA segments leading to nucleosome structure changes and exposing or obscuring of DNA to gene regulatory factors. These in turn lead to changes in transcription and translation of encoding DNA and protein synthesis (Fig. 34.2). As an example, acetylation of lysine residues of histones opens up the protein-encoding-DNA chain and leads to increase of transcription. Recent studies show that inherited or acquired epigenetic alterations and dysregulations are in close relation to many diseases particularly malignancies. An example of epigenetic changes is the indirect effect of mutant genes of isocitrate dehydrogenase (IDH) enzymes and their relation with the formation of chondromas (Table 34.1). Many of the epigenetic alterations and dysregulations are reversible. Thus, diseases that occur on the base of these dysregulations could be treatable [2, 7–9].
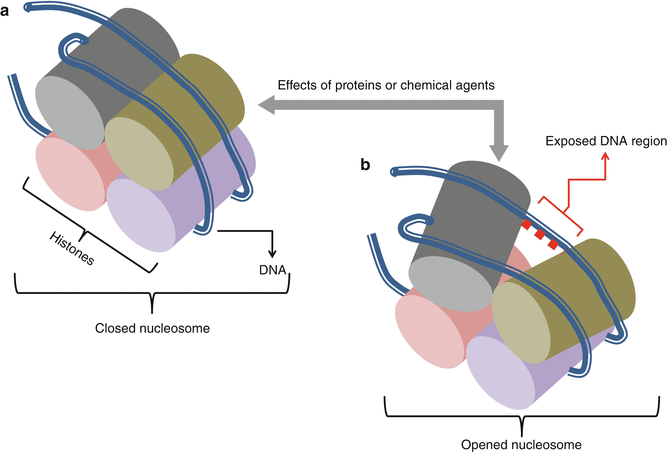
Fig. 34.2
Changes in the form and compactness of nucleosomes as epigenetic factors of gene expression. (a) closed (b) opened nucleosome under the effects of proteins or chemical agents
Other than nuclear located events another important site of modulation of the cell metabolism and functions is mitochondria. These cytoplasmic organelles have their own DNAs although in small amounts. They can perform their own DNA replication, transcription, and translation. Mitochondria have important roles in cell energy generation, anabolic metabolism, and apoptosis. These functions of mitochondria can be regulated due to the necessities of the cell. Unlike the normal cells, in rapidly growing cells the main function of mitochondria change from energy generation into promotion of synthesis of basic molecules to be used in the structure of cellular components. Mitochondrial DNA, different from nuclear DNA, is inherited maternally. But as mitochondrial proteins can be produced by both nuclear and mitochondrial DNA, genetic diseases in association with mitochondrial dysfunctions can be either maternally inherited, X-linked, or autosomal [2, 10, 11].
Apoptosis or programmed cell death is an important mechanism of both normal and tumor tissue maintenance and growth. In normal tissue, apoptosis is initiated by sensation of irreparable cell abnormalities. These abnormalities might concern any one of cellular events. Genetic alterations responsible for oncogenesis also initiate apoptosis. The mechanism of apoptosis is triggered both by intrinsic and extrinsic pathways. Extrinsic pathway is driven by the cell signaling system and will be explained in the next paragraph. Intrinsic pathway works through mitochondrial functions. Intrinsic signals like DNA damage or inadequate synthesis of vital proteins lead to mitochondrial dysfunction. Consequently, mitochondrial contents leak out into the cytoplasm. Some of these contents form complexes by cytoplasmic proteins, which in turn activate apoptotic enzymes. Failure of apoptosis is one of the basic mechanisms of and requirements for the development of malignancies [1–3].
Cell signaling and communications are indispensable features for growth, differentiation, liveliness, proliferation, and apoptosis of the cells and also maintenance of optimal organization of the concerning tissues. These events are controlled by numerous and various signals like ions, growth factors, hormones, and cytokines. Exposure of the cell to the signals is a continuous event. When a cell is exposed to insufficient appropriate signals apoptosis occurs. Signaling molecules or combinations of them ultimately initiate specific cell biological responses. In brief, signals are perceived by the cells through specific receptor proteins that are located over their surfaces. Activated receptor proteins in turn lead to reproduction of active transcription factors through several types of intracellular intermediate pathways. At the end, transcription factors enter nucleus and alter related gene expression. Mutations or alterations of the genes that encode proteins for this pathway might lead to over-inducing or inhibiting of the cell signaling mechanisms. These abnormalities have roles in unregulated cell proliferation and occurrence of cancers. Among the cell signal agents, growth factors and their receptors have the most important roles in oncogenesis. In normal conditions these protein-made factors stimulate cell events like growth, synthesis of cellular components, migration, differentiation, and proliferation. These effects of growth factors are mediated through their specific receptors to enhance or maintain normal tissue balance or to replace injured tissue regions. Dysregulation of the cell signaling pathways due to abnormal growth factors can lead to uncontrolled cell proliferation. Alterations of growth factor genes, leading to over-expression, have roles in several types of cancers. An example is over-expression of platelet-derived growth factor (PDGF)-beta gene and its relation by formation of dermatofibrosarcoma protuberans (Table 34.2) [1–3].
Cells, normal or abnormal, should proliferate to maintain or increase their population. Proliferation of the cells comes true by mitosis of each divisible cell into two daughter ones through the process of cell cycle. Cell cycle has four phases and three control points (Fig. 34.3). Its phases are G1 (initial or presynthetic growth), S (DNA replication and synthesis), G2 (gap or premitotic growth), and M (mitosis). When a cell is not genetically active and has no tendency to be divided, it is in stable or quiescent phase which is shown by G0. Cell cycle can begin both by activation of a G0 quiescent cell or by direct entrance of a just formed daughter cell into a new cycle. This occurs due to the type and necessities of the tissue. Cell cycle is controlled by a network of proteins and enzymes that are appointed one after the other (Fig. 34.4). Regulators of the cell cycle are cyclin-dependent serine/threonine kinase (CDK) named enzymes that are activated by binding to appropriate cyclin named proteins. Cyclin-CDK complexes are phosphorylated and activated by cyclin-activating kinases (CAKs). These activated complexes then, in turn, phosphorylate and activate control point protein components that are necessary for the progression of cell cycle. Two of the most important control point protein components are retinoblastoma (Rb) and p53 proteins. On the other hand, CDK-inhibitors (CDKIs) silence CDKs and halt the cell cycle through the control points. CDK-inhibitors are proteins that are shortly shown by p and a number (i.e., p16). The main aim of the cell cycle control points is fixing probable DNA irregularities. Control points of cell cycle are composed of one restriction point and two checkpoints (Fig. 34.3). The restriction point is located at the G1 phase. After passing this point, the cell would not respond to the removal of the growth factors. Checkpoints are located at the G1/S and G2/M interphases of the cycle (Fig. 34.3). The G1/S checkpoint is the primary location for fixation of the DNA irregularities before the expenditure of the cellular sources for dividing process. When this checkpoint is passed, the cell is forced to complete mitosis. At the G2/M checkpoint, damage or appropriateness of the duplicated DNA is controlled. Mutations of the genes that encode CDKs, cyclins, CDKIs, or control point protein components have roles in the development of cancers when they create a net effect of promoted progression of cell cycle and uncontrolled cell proliferation. Amplifications of CDK and cyclin genes facilitate progression of the cell cycle through checkpoints. The most frequent type of CDK mutation occurs in CDK4 genes. Over-expressed CDK4 binds to D cyclin and facilitates progression of cell cycle through G1 checkpoint by phosphorylation of Rb. Mutation and over-expression of CDK4 is one of the genetic alterations that are in association with osteosarcoma (Table 34.2). Mutations in CDKI, p53, and Rb genes that interrupt the functions of their related proteins lead to the increased tendency of oncogenesis through ineffective cell cycle checkpoints (Table 34.1) [1–3].
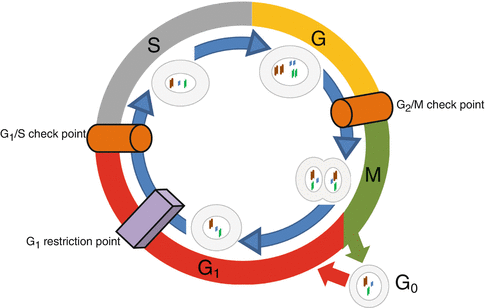
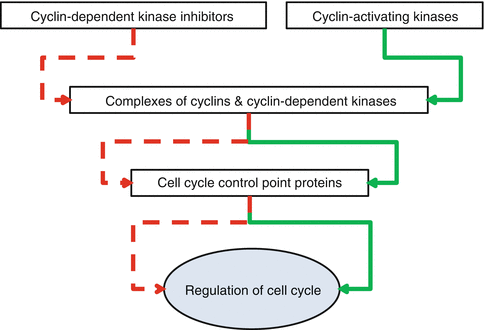
Fig. 34.3
Cell cycle phases and control points
Fig. 34.4
Layers of protein network responsible for the regulation of cell cycle. Inhibitor effect is shown by red and activator effect by green arrows
Molecular Basis of Oncogenesis
Cancers are expressions of uncontrolled multiplying of the cells due to aberrations in their genetic contents. To be able to lead to cancer, genetic aberrations should not be lethal for the precursor cells and could be passed to their daughters. These aberrations should interest one of the growth-inducing (proto-oncogene), growth-inhibiting (anti-oncogene), apoptosis-regulating, or DNA-repairing genes. In normal conditions, all of the cell events are under the harmonious effects of these genes. Oncogenesis occurs when the balancing effects of these genes is impaired. Actually, any cancer does not occur due to a single gene mutation. New cell generations derived from mutant precursor cells require subsequent accumulating mutations to produce a clinically evident cancer (Fig. 34.5). The number of these subsequent mutations differs between dozens to even thousands. As DNA-repairing processes are the first step of body mechanisms against the formation of cancers, gene mutations that result in loss of DNA-repairing functions are the main predisposing factors of cancer formation (Table 34.1). These mutations lead to genomic instability without phenotypic change in related cells. With time, these cells acquire several accumulating mutations. Due to myriad numbers of possible genetic and epigenetic alterations, genotypes of tumor cells even in phenotypically similar tumors show significant differences. Despite the genotypic diversity, all the cancer cells are subjected to indispensable common physiological changes. These changes have been described in 2000 and revised in 2011 as hallmarks of the cancer by Hanahan and Weinberg. In the following paragraphs, molecular and biochemical bases of oncogenesis are described regarding each one of these standard hallmarks [3, 12–16].
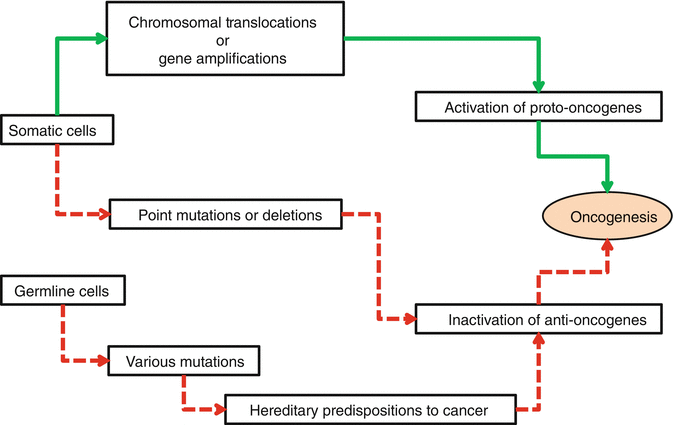
Fig. 34.5
Schematic diagram of subsequent accumulating mutations that are responsible for oncogenesis. Green arrows represent the mutation of proto-oncogenes and red arrows represent mutations of anti-oncogenes
The first hallmark is proliferation capacity without the need of inducing factors from outside of the tumor. This means that tumor cells can produce their own signals for growth. This property is usually gained by proto-oncogene mutations. Proto-oncogenes function as inducers of cell growth and proliferation. Dominant alterations of them produced by mutations are named oncogenes. As mentioned previously, the main mechanisms of over-expression of oncogenes are based on chromosomal translocations and gene amplifications. Over-expressed oncogenes encode oncoproteins that have the ability to induce the cell growth without responding to cell cycle checkpoints. Genetic alterations that lead to over-expression of proto-oncogenes in association with musculoskeletal tumors are summarized in Table 34.2 [3, 14–23].
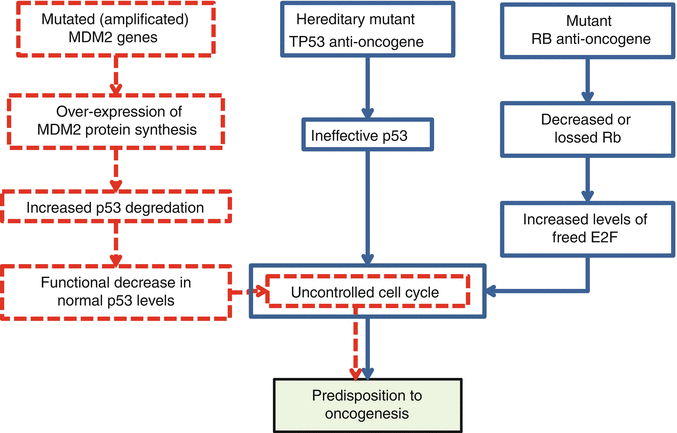
The second hallmark is unresponsiveness of the tumor cells to growth inhibitory signals. Proteins that take part in inhibition of the cell growth process are called tumor suppressive proteins and are coded by anti-oncogenes. In normal conditions, all of the tumor suppressive proteins work harmoniously as the safe brake system of the cell cycle. In the case of abnormality or insufficiency, inhibitory mechanisms of cell proliferation halt. These create tendency and predisposition to oncogenesis due to failure or decrease in the responsiveness of the tumor cells to growth inhibitory signals (Fig. 34.6). This hallmark is a functional loss and usually is gained due to biallelic recessive mutations of anti-oncogenes. As mentioned previously, the most frequent types of anti-oncogene mutations are point mutations and deletions. Alterations of anti-oncogenes that are in relation with increased risk of formation of musculoskeletal tumors are summarized in Table 34.1. In musculoskeletal system tumors, frequent genetic alterations that lead directly to the hallmark of unresponsiveness to the growth inhibitory signals involve TP53 and RB anti-oncogenes (Table 34.1 and Fig. 34.6). Frequently seen indirect mechanism of this hallmark occurs through over-expression of MDM2 gene (Table 34.2 and Fig. 34.6) [3, 14–16, 18–22, 24–26].
 Stem Cell Therapy and Orthopedics
Stem Cell Therapy and Orthopedics
 Nonsteroidal Anti-inflammatory Drugs (NSAIDs) and Corticosteroids
Nonsteroidal Anti-inflammatory Drugs (NSAIDs) and Corticosteroids
 Fundamentals of Quantitative Gait Analysis
Fundamentals of Quantitative Gait Analysis
 Skeletal Muscle; Structure, Function, and Repair
Skeletal Muscle; Structure, Function, and Repair
 Lower Extremity
Lower Extremity
 Nuclear Medicine Techniques in the Diagnosis and Treatment of Diseases of the Musculoskeletal System
Nuclear Medicine Techniques in the Diagnosis and Treatment of Diseases of the Musculoskeletal System




Fig. 34.6
Direct (blue flowcharts) and indirect (red flowchart) mechanisms of predisposition to oncogenesis
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:
Stem Cell Therapy and Orthopedics
Nonsteroidal Anti-inflammatory Drugs (NSAIDs) and Corticosteroids
Fundamentals of Quantitative Gait Analysis
Skeletal Muscle; Structure, Function, and Repair
Lower Extremity
Nuclear Medicine Techniques in the Diagnosis and Treatment of Diseases of the Musculoskeletal System
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
